







Atrial fibrillation (AF) is the most common arrhythmia experienced in clinical practice, and is responsible for significant morbidity and mortality.1 It affects more than 6 million people in Europe.1 The lifetime risk of developing AF after the age of 40 is approximately 25 %.1 AF is a major public health burden as it is associated with an increased risk of stroke by fivefold, dementia by twofold, heart failure by threefold and mortality by twofold.1,2
Current therapeutic options include non-invasive treatment using antiarrhythmic agents and invasive methods using catheter ablation.3,4 Although several improvements have been achieved, challenges still remain. Pulmonary vein isolation is now an established treatment, especially for patients with paroxysmal AF, but success rates (freedom from AF) in persistent AF are still insufficient.3,5,6 To further improve AF treatment, mechanisms underlying AF initiation and maintenance have to be further elucidated to better enable identification of the most suitable treatment strategy for each individual patient (in terms of drug toxicity, freedom of AF symptoms and improved quality of life) and recognition of patients at high risk for AF.
In recent years our understanding of AF pathophysiology has markedly improved by the identification of key players in cardiovascular signalling: microRNAs (miRNAs).7 We will provide a comprehensive overview of the role of miRNAs in AF and their potential therapeutic implications.
Pathophysiology of Atrial Fibrillation
The pathophysiological basis of AF is multifactorial and complex.8,9 Electrophysiological hallmarks of AF are ectopic activity and reentry as the functional surrogates for trigger and susceptible substrate. Underlying molecular mechanisms include changes in expression and function of ion channels, altered calcium homeostasis, enhanced atrial automaticity, alterations in gap junction distribution, and adverse effects on atrial integrity such as dilatation, fibrosis or inflammation. These mechanisms are summarised as electrical and structural remodelling, and will be described here briefly.
Three main mechanisms causing focal ectopic activity are: enhanced atrial automaticity, early afterdepolarisations and delayed afterdepolarisations. Under normal physiological conditions, generation and conduction of electrical impulses in the heart is implemented by a characteristic sequence of voltage changes driven by depolarising and repolarising ion currents. Cardiomyocytes display a resting membrane potential of around -80 mV. An electrical impulse causes a swift depolarisation by rapidly activated sodium channels with subsequent influx of sodium ions. Following this, potassium ions exit the cell through potassium channels, initiating the repolarisation of the cell (re-establishing the resting membrane potential). Simultaneously, calcium ions enter the cell leading to cell contraction (excitation– contraction coupling) and slowing of the repolarisation.
In healthy hearts the sinus node dominates the generation of electrical signals. However, it is possible that a cell outside the sinus node reaches the threshold potential earlier resulting in ectopic firing at a more rapid rate, potentially leading to atrial tachycardia. In this regard alterations of the cellular calcium homeostasis may play an important role as has been demonstrated in animal models and patients with AF.9 Delayed afterdepolarisations result from an abnormal calcium leak from the sarcoplasmatic reticulum (SR). Physiologically, cellular calcium is removed by the SR Ca2+ATPase (SERCA) and the Na+–Ca2+ exchanger (NCX) during diastole to re-establish ionic homeostasis at the end of the cardiac cycle. During atrial tachycardia, however, calcium is progressively accumulated in the cell because of repetitive activation of the L-type Ca2+ channel. This Ca2+ overload leads to multiple maladaptive alteration, for example, a Ca2+ overload of the SR with concomitant dysfunction of the ryanodine receptor (SR calciumrelease channel). This distribution of the SR regulation can cause spontaneous diastolic Ca2+ release from the SR during diastole, which then can activate the electrogenic NCX (Ca2+ versus 3 Na+) causing a depolarising inward current (delayed afterdepolarisation). This leads to a progressive depolarisation of the cell and ectopic firing (when the threshold potential is reached; so-called triggered activity).10–12 Early afterdepolarisations occur when the action potential duration (APD) is excessively prolonged, for example, in the context of arrhythmia syndromes such as long-QT syndrome.9,11,12 In this setting, another phenomenon called dispersion of repolarisation has been described. In healthy myocardia, a more or less homogeneous-organised repolarisation prevents onset of arrhythmia; in diseased myocardium, however, a vulnerable substrate can be created by heterogeneous electrophysiological properties due to remodelling processes. This may be caused by transmural APD variations within the 3D myocardial structure. These changes causing electrophysiological heterogeneity can result in proarrhythmic repolarisation differences predisposing to the initiation of arrhythmias. Pacemaker cells express specific ion channels (funny channels) that are responsible for the so-called automaticity (i.e. the progressive diastolic depolarisation). Therefore, an upregulation of these ion channels, as seen in heart failure, could be a possible mechanism leading to enhanced automaticity.13
Another hallmark of AF is reentry that can occur when at least two (functionally or anatomically) distinct pathways, a unidirectional block in one of the pathways and a slowed conduction, are present. In this case the conduction time along the unblocked pathway must exceed the refractory period of the blocked pathway (conduction time x reentry circle length > refractory period). In healthy myocardia, the electrical properties are relatively homogeneous without slow conduction areas preventing the occurrence of reentry. In diseased myocardia, however, two mechanisms are important: altered electrical properties that cause a shortening of the refractory period or conduction slowing, and structural changes (e.g. atrial fibrosis) that disturb the uniform and homogeneous excitation and thereby provide an anatomical substrate for reentry. In a healthy heart, reentry cannot occur because cardiomyocytes display a certain refractory period that prevents premature stimuli to conduct. When the refractory period is shortened, however, the cell is excitable earlier, ectopic activity can be conducted and reentry can be initiated. Additionally, slowed conduction velocity can also allow reentry because cells are excitable again when the impulse arrives. Structural changes in the atrium such as dilatation and fibrosis extend conduction pathways, slow conduction and create conduction barriers favouring initiation and maintenance of reentry circuits. Interestingly, some of the electroanatomical changes implicated in AF pathophysiology initially act as a protective mechanism of the cell, but finally result in a fixed substrate for AF maintenance. Atrial tachycardia (as seen in AF) causes a cellular calcium overload. To reduce the calcium influx and to antagonise the calcium overload, the calcium current is reduced (by inactivation of calcium channels (short-term effect) and reduced gene expression of the calcium channels (long-term effects), leading to a shortening of the action potential. This favours reentry and therefore contributes to AF maintenance (‘AF begets AF’).14
MicroRNAs as Novel Regulators of Cardiac Arrhythmogenic Remodelling
In 1993 it was discovered that development of Caenorhabditis elegans is regulated by a short RNA fragment named Lin-4 that inhibits the expression of lin-14 by binding to the 3’ untranslated region of its mRNA.15,16 In the following years, these miRNAs were identified as key players in molecular signalling leading to disease. Although other noncoding RNA species have been discovered so far,17,18 we will focus on miRNAs in this review.
For a detailed description, especially of miRNA biogenesis we refer to previously published reviews.19–22 Briefly, miRNAs are short (approximately 22 base pairs), single-stranded, non-coding RNAs that regulate post-transcriptional gene expression. MiRNAs can induce down- and upregulation of genes either by direct inhibition of their target mRNA (causing direct downregulation of their target gene) or inhibition of an endogenous antagonist of another gene (causing indirect upregulation of a gene dependent on the miRNA’s target gene). MiRNAs are broadly conserved among species; to date, thousands of miRNAs have been discovered in plants, insects and mammals. According to the latest release of the online database miRBase (version 21.0, June 2014) 2,588 mature miRNAs have been identified in humans.23 Each miRNA can act on several target genes and each gene can be affected by several miRNAs. This complex regulatory network results in up- and downregulation of agonists and antagonists at the same time leading to the idea of miRNAs as fine tuners of gene expression.24
Experiments on genetic ablation of the enzyme dicer that is one of the key enzymes in miRNA biogenesis, revealed the crucial role for miRNAs in normal development as mice and zebrafish with dicer knockout are not viable.25 A cardiac-specific knockout of dicer in mice also resulted in premature lethality due to cardiac dilatation and heart failure.26 Finally, dicer is essential even in the adult organism as shown by da Costa Martins et al., who demonstrated that a reduced cardiac dicer activity leads to increased incidence of sudden cardiac death, cardiac hypertrophy and expressional switch from an adult to a fetal gene expression programme.27
These studies offered compelling evidence that microRNAs play an important role in normal cardiac development and disease.26,27 Therefore, a growing number of studies has been published evaluating the role of miRNAs in cardiac electrical and structural remodeling, potentially contributing to the initiation and maintenance of AF pathophysiology.7,9
miRNAs Involved in Electrical Remodelling
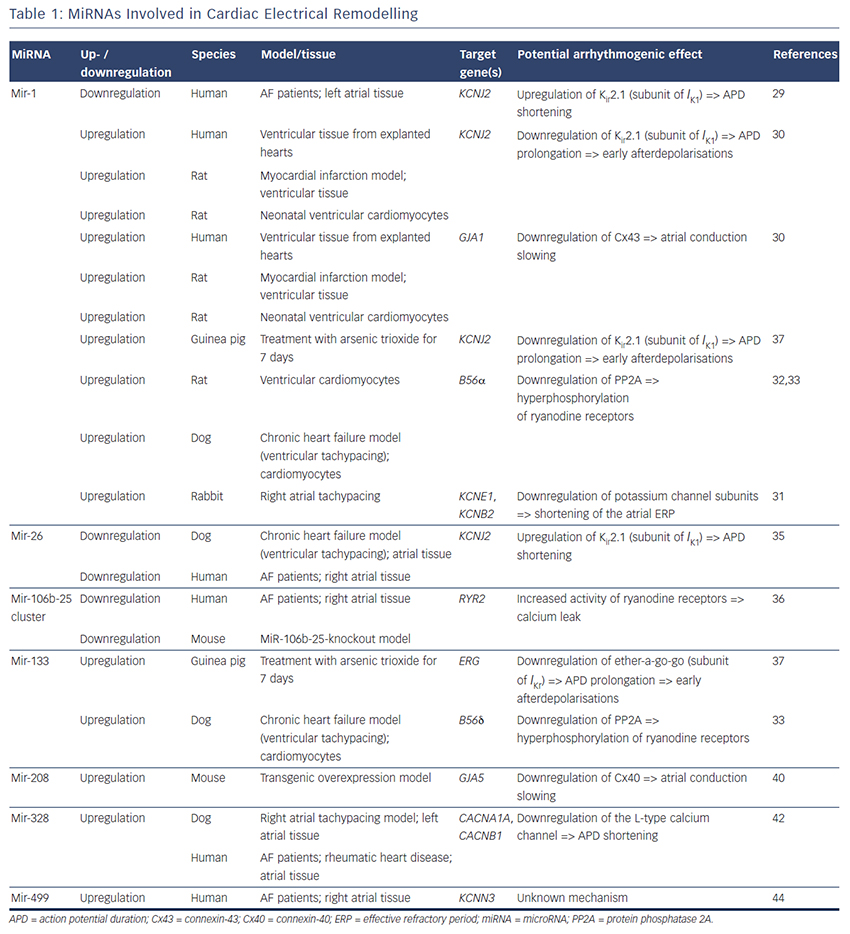
MiRNAs involved in cardiac electrical remodelling are miR-1, miR-26, miR-208a, miR-328 and miR-499 (Table 1, Figure 1). Their target genes are encoding ion channels, connexins or proteins involved in calcium signalling resulting in conduction slowing or shortening of the action potential duration, which are hallmarks of AF pathophysiology.
miR-1
MiR-1 and miR-133 are muscle-specific (skeletal and heart) miRNAs that are expressed from bicistronic clusters and, therefore, are among the most abundant miRNAs in the heart.28 Girmatsion et al. showed that miR-1 is downregulated in the heart of patients with persistent AF compared with patients with sinus rhythm.29 They demonstrated that this downregulation of miR-1 is accompanied by a significant upregulation of its target gene KCNJ2 that encodes Kir2.1, the subunit of the inward rectifier potassium channel IK1. Additionally, they showed that an ex vivo tachystimulation of atrial tissue resulted in downregulation of miR-1 and upregulation of Kir2.1 protein and IK1 density. An increased IK1 density is associated with a shortening of the APD and could therefore enable reentry and AF maintenance.
Yang et al. indicated another role for miR-1 in arrhythmogenesis as they confirmed KCNJ2 as an miR-1 target gene, but also identified GJA1 as an additional target gene that encodes connexin-43.30 They performed a miR-1 overexpression in an ischaemic rat model (which is in contrast to the miR-1 downregulation in patients with AF). Overexpressing miR-1 in the rat model led to downregulation of KCNJ2 and GJA1 resulting in APD prolongation and conduction slowing.
In 2013, Jia and colleagues published a study evaluating the role of miR-1 in rabbits.31 They performed right atrial tachypacing in rabbits for 1 week resulting in increased inducibility of AF due to a shortening of the atrial effective refractory period and an increase in the slowly activating delayed rectifier potassium current (IKs). They found an upregulation of miR-1 paralleled by a downregulation of KCNE1 (coding for the voltage-gated potassium channel subfamily E member 1, minK) and KCNB2 (coding for the voltage-gated potassium channel subfamily B member 2, Kv2.2). Using anti-miR-1 inhibitor oligonucleotides, they rescued the phenotype and prevented expressional changes of miR-1, KCNE1 or KCNB2.
Additionally, miR-1 is implicated in playing a role in calcium homeostasis. In rat cardiomyocytes, Terentyev et al. induced an overexpression of miR-1 resulting in downregulation of its target B56α, a regulatory subunit of the protein phosphatase 2A (PP2A).32 PP2A now dissociates from the ryanodine receptor leading to hyperphosphorylation of this calcium channel. Hyperphosphorylated ryanodine receptors show arrhythmogenic spontaneous calcium release that can cause afterdepolarisations.32,33
miR-26
MiR-26 has also been shown to regulate gene expression of KCNJ2.34,35 In a canine AF model and in human atrial samples, miR- 26 is downregulated while Kir2.1 is upregulated. Overexpression of miR-26 resulted in suppressed KCNJ2 expression, miR-26 knockdown resulted in enhanced KCNJ2 expression. These results were confirmed in a mouse model performing virus-induced miR-26 overexpression and miR-mask induced miR-26 knockdown:35 reduced/enhanced AF vulnerability was observed in miR-26 overexpression/knockdown.
miR-106b-25 cluster
Recently, Chiang and colleagues identified the miR-106b-25 cluster (miR-25, miR-93 and miR-106b) as mediators of electrical remodelling.36 They showed a downregulation in the atria of patients with AF and found an increased susceptibility for AF in miR-106b-25-knockout mice due to increased SR calcium release (SR calcium leak) mediated by an enhanced ryanodine receptor expression (which is the confirmed target gene of miR-93).
miR-133
Shan and colleagues evaluated the role of miR-133 on the QT interval in a guinea pig model.37 An upregulation of miR-133 resulted in significantly decreased protein levels of ERG, a subunit of the potassium channel responsible for the delayed rectifier potassium current Ikr, accompanied by a prolonged QTc interval and increased mortality rates. These findings were reproduced by direct application of miR-133 while miR-133 blockade using an antisense inhibitor abolished the effects. Interestingly, they also confirmed previous results of miR-1 in their model (upregulation of miR-1, downregulation of Kir2.1 protein).37
Matkovich et al. generated a miR-133 overexpressing mouse that displayed QT/APD prolongation and a reduced outward potassium current Ito,f.38 Although they could measure a reduced expression of the Ito,f subunit KCNIP2, they did not validate this gene as a miR-133 target.
Belevych et al. recently published a study in a canine model of heart failure.33 They demonstrated that miR-133 is upregulated leading to downregulation of its target gene B56δ, a catalytic subunit of PP2A with similar effects as B56α (that is downregulated as a target gene of miR-1, see above).
miR-208a
MiR-208 is an interesting miRNA as the two isoforms (miR-208a and miR-208b) are differentially expressed during cardiogenesis.39 MiR- 208a is encoded within an intron of the α-cardiac muscle myosin heavy chain gene MYH6 (adult isoform), whereas miR-208b is encoded within an intron of the β-cardiac muscle myosin heavy chain gene MYH7 (fetal isoform). MiR-208 isoforms are expressed along with their host genes: miR-208b is expressed during cardiogenesis while miR- 208a is expressed in adult hearts. Interestingly, pathological cardiac remodelling is associated with the induction of a fetal gene expression pattern with re-expression of MYH7 and, therefore, miR-208b.40,41 To evaluate miR-208 in cardiac remodeling, Callis et al. performed an overexpression of miR-208a in a mouse model and observed an increased vulnerability towards arrhythmias.40 They demonstrated that miR-208 targets GJA5 encoding the cardiac gap junction protein connexin-40 and therefore mediates pro-arrhythmogenic remodelling by altering gap junction expression.
miR-328
Another miRNA involved in calcium signalling is miR-328. It is upregulated in atrial tissue of patients with AF and in a canine AF model.42 CACNA1C and CACNB1, subunits of the L-type calcium channel, were identified as target genes of miR-328. In the canine model, an adenovirus-induced miR-328 overexpression caused reduced L-type calcium current and APD shortening and increased AF vulnerability. Knockdown of miR-328 with an antagomiR reversed these effects suggesting a potential role for miR-328 in cardiac electrical remodelling via the L-type calcium channel.
miR-499
KCNN3 encodes the calcium-activated potassium channel 3 (SK3) and is potentially involved in AF pathophysiology as common genetic variants within this gene are associated with AF.43 Recently, Ling and co-workers reported that miR-499 was elevated in human atrial tissue; SK3 protein expression, however, was downregulated.44 Finally, they provided evidence of a direct interaction between miR-499 and KCNN3 by in vitro overexpression and knockdown experiments. However, the authors analysed only eight patients (four with AF, four without AF [controls]) and did not report any enhanced arrhythmogenity in vitro or in vivo. Thus, the functional role of SK3 channels in AF pathophysiology still remains unclear as overexpression of SK3 channels in mice did not result in development of AF, but in an increased incidence of sudden death due to bradyarrhythmias and heart block.45
MiRNAs Involved in Structural Remodelling
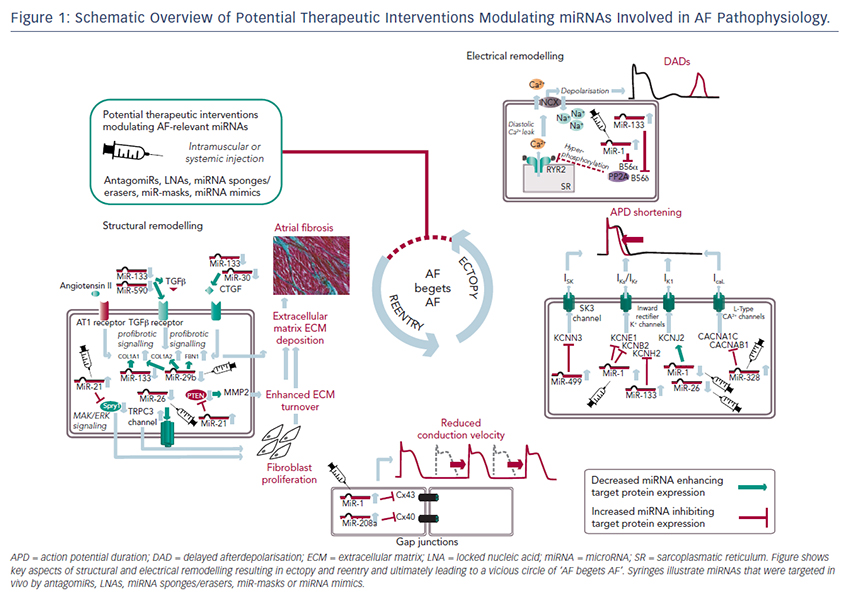
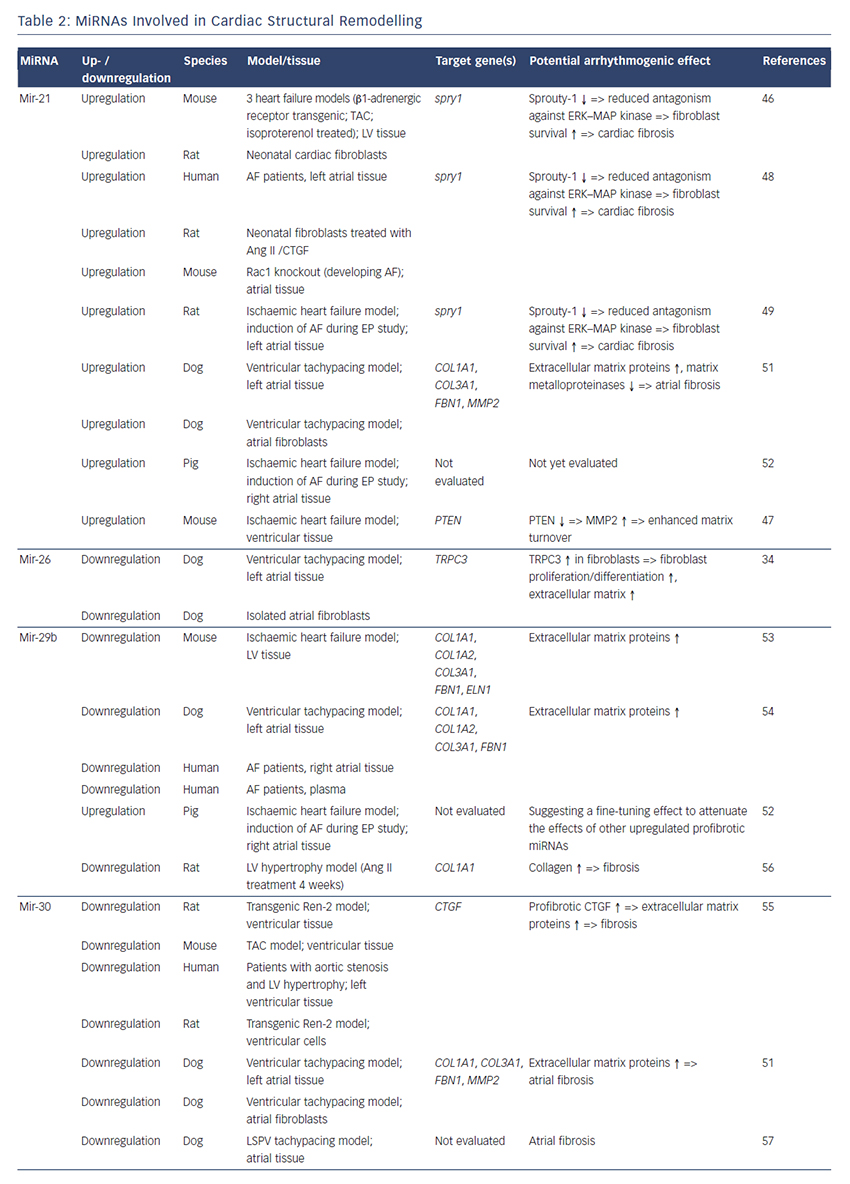
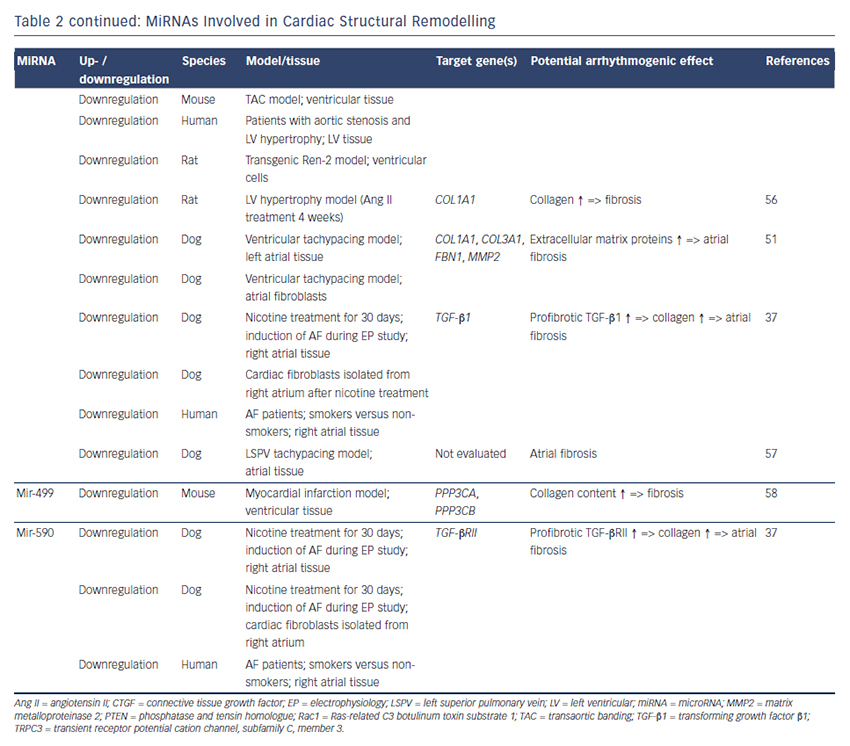
MiRNAs involved in cardiac structural remodelling are miR-21, miR- 26, miR-29b, miR-30, miR-133 and miR-590 (Table 2, Figure 1). These miRNAs regulate genes encoding proteins that are involved in extracellular matrix turnover and pro- or antifibrotic signalling cascades leading to atrial fibrosis as the anatomical substrate for reentry.
miR-21
A first report of miR-21 being involved in profibrotic signalling was published by Thum et al. in 2008.46 In a murine heart failure model, they showed that miR-21 is upregulated mostly in cardiac fibroblasts. By targeting sprouty homologue 1 (Spry1) miR-21 regulated the profibrotic ERK–MAP kinase signalling pathway. In vivo knockdown of miR-attenuates cardiac fibrosis and improves cardiac function.
In a mouse model of myocardial infarction (MI), miR-21 was upregulated leading to repression of the transcription factor phosphatase and tensin homologue (PTEN).47 This blockade results in an upregulation of matrix metalloproteinase 2 (MMP2) and an enhanced matrix turnover suggesting a role for miR-21 in ventricular remodelling.
Finally, miR-21 was detected to be upregulated in left atrial tissue of patients with AF.48 This was associated with reduced expression of Spry1 and increased expression of the profibrotic connective tissue growth factor (CTGF), lysyl oxidase and Rac1-GTPase, resulting in increased collagen content. Additionally, it could be shown in vitro that CTGF and angiotensin II induce an upregulation of miR-21. Finally, Adam and co-workers generated a Rac1-GTPase-knockout mouse and confirmed their previous results in that animal model suggesting a potential mechanism on the profibrotic action of angiotensin II via miR-21.
Cardin et al. showed that miR-21 is upregulated in an ischaemic heart failure model in rats.49 Again, Spry1 was confirmed as the miR-21 target gene being downregulated. They injected a miR-21 antagonist directly into the left atria resulting in significant knockdown of miR-21, upregulation of Spry1, improvement of myocardial function, reduction of atrial fibrosis and, finally, reduced AF duration.
Our group evaluated the role of several miRNAs in atrial remodelling in a canine AF model, in which heart failure is induced by ventricular tachypacing, leading to a subsequent atrial remodelling with increased susceptibility for AF.50 We showed that tachycardiomyopathy is associated with a significant upregulation of extracellular matrix proteins (collagen-1, collagen-3 and fibrillin) and a downregulation of MMP2 in left atrial tissue as well as in cardiac fibroblasts.51 These changes were accompanied by increased atrial fibrosis and an upregulation of miR-21. Other miRNAs that are discussed in detail below (miR-29b, miR-30a, miR-133a) were decreased in our model.
Because tachycardiomyopathy is rare in humans we also established an ischaemic heart failure model in pigs to evaluate proarrhythmogenic atrial remodelling processes.52 We induced MI by balloon occlusion of the left anterior descending artery. After 4 weeks of reperfusion, pigs showed a significant heart failure phenotype (left ventricular [LV] pressure increased, LV ejection fraction reduced, LV angiography reduced), significant atrial fibrosis (left and right atria) and were more prone to AF than healthy control pigs (AF inducibility increased, AF burden increased, AF duration/induction increased). In our first analysis, we demonstrated a significant upregulation of miR-21, which accompanied the fibrotic remodelling of the atria.52
In summary, several reports provide consistent evidence regarding a potential role of miR-21 on atrial structural remodelling and AF pathophysiology.
miR-26
Besides its role in electrical remodelling (described above), miR-26 is also reported to be involved in structural remodelling.34 In the canine ventricular tachypacing (VTP) model, miR-26 was downregulated in fibrillating atria causing an upregulation of transient receptor potential cation 3 (TRPC3) channels. These TRPC3 channels have been shown to be expressed in cardiac fibroblasts regulating calcium influx, cell proliferation, extracellular signal-regulated kinase phosphorylation and α-smooth muscle actin protein expression. In vivo blockade of TRPC3 channels prevented the development of an AF substrate in the canine VTP model.
miR-29b
In 2008 van Rooij et al. reported a downregulation of miR-29b in a mouse model of MI that was accompanied with increased fibrosis and an upregulation of extracellular matrix proteins as collagen, fibrillin and elastin.53 They could reproduced their results in vitro by miR-29b overexpression and knockdown, respectively.
Our group evaluated the role of miR-29b in atrial remodelling in the canine VTP model.50 After 24 hours of tachypacing, miR-29b was significantly downregulated in atrial tissue as well as in atrial fibroblasts and remained decreased throughout the time course (up to 2 weeks of ventricular pacing).54 Extracellular matrix proteins (collagen, fibrillin) were upregulated, atrial fibrosis was enhanced and the induced AF duration was significantly prolonged. Lentiviral miR-29b manipulation in fibroblasts mimicked the effects seen in the canine model: overexpression of miR-29b led to a downregulation and miR- 29b knockdown led to an upregulation of extracellular matrix proteins. Interestingly, we confirmed these changes (miR-29b downregulation) in right atrial tissue of patients with AF.
Other miRNAs Involved in Structural Remodelling
Duisters et al. analysed ventricular tissue from rats (hypertensioninduced heart failure model) and mice (transaortic banding [TAC] model), rat cardiac cells and human ventricular biopsy samples (patients suffering from aortic stenosis with LV hypertrophy versus patients who have had coronary artery bypass grafting without LV hypertrophy).55 Their results were consistent over species: miR- 30c and miR-133 were downregulated, whereas their target gene CTGF, a profibrotic mediator, was upregulated. In vitro manipulation achieving miRNA overexpression/knockdown confirmed their results and provided evidence for a potential role of these miRNAs in structural remodelling.
Castoldi et al. injected angiotensin II in rats for 4 weeks resulting in significant hypertrophy and fibrosis.56 They showed a downregulation of miR-133 and miR-29b paralleled by upregulation of collagen-1. All the effects were abolished when rats were treated with irbesartan, suggesting a role for miR-133/miR-29b in angiotensin-II induced cardiac remodelling.
Besides these studies on ventricular hypertrophy, our group identified an atrial downregulation of miR-30 and miR-133 in the canine VTP model accompanied by increased atrial fibrosis.51 This finding was confirmed by Li et al. who induced AF (without heart failure) in dogs by rapid pacing of the left superior pulmonary vein for 5 weeks.57
In an MI model in mice, Wang and co-workers observed a downregulation of miR-499 in the area at risk after ischaemia.58 For a further analysis, they produced a miR-499 transgenic mouse and observed improved cardiac function and reduced collagen content after MI. The catalytic subunits of calcineurin (PPP3CA, PPP3CB) were identified as target genes of miR-499. Further analysis revealed alterations in mitochondrial fission and apoptosis signalling as the potential mechanism underlying the miR-499 actions.
Atrial remodelling was further investigated by Shan et al.37 They administered nicotine in dogs for 30 days resulting in significant increased atrial fibrosis and AF vulnerability compared with healthy dogs. The profibrotic mediator transforming growth factor b 1 (TGF- β1) and the TGF-β receptor-II (TGF-βRII) were significantly upregulated, whereas miR-133 and miR-590 were downregulated in the right atrium. They identified TGF-β1 as the target gene of miR-133 and TGF-βRII as the target gene of miR-590. In vitro manipulation confirmed their in vivo results (up-/downregulation of miR-133/miR-590 resulted in down-/ upregulation of TGF-β1/TGF-βRII/collagen). Finally, they examined right atrial tissue of AF patients and could show that miR-133/miR-590 were downregulated in smokers, suggesting a potential mechanism of the increased risk of AF in smokers.
MiRNAs as Potential Therapeutics
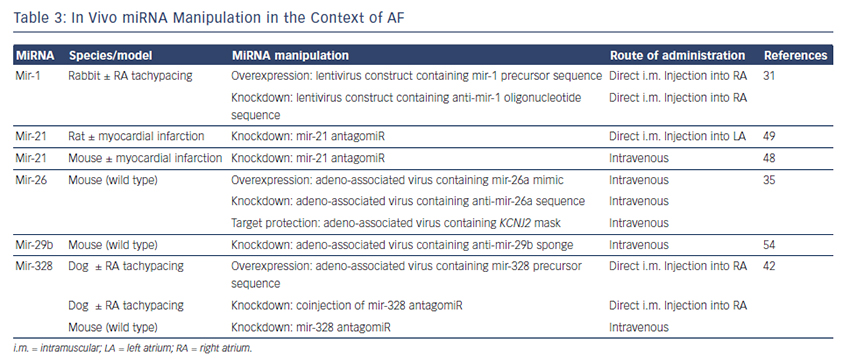
The findings described above demonstrate compelling evidence that miRNAs are powerful factors in AF pathophysiology suggesting in vivo manipulation for AF treatment. So far, several options to agonise or antagonise miRNA effects were developed and successfully evaluated in vivo in AF-related animal models, as described above and summarised in Table 3.31,35,42,48,49,54
Overexpression of a miRNA that is downregulated in disease can be achieved by miRNA mimics. Mimics are synthetic double-stranded RNAs that are incorporated and processed by the cell-like endogenous miRNAs and therefore ‘mimic’ their effects.59 However, mimics are not tissue- or cell-type specific and can therefore create undesirable offtarget effects. This can be avoided by using cardiotropic adenoassociated virus-mediated miRNA transfer that has been shown for the treatment of heart failure in mice60 and cardiac hypertrophy in rats.61
For antagonising a pathological miRNA upregulation, several knockdown approaches are available including anti-miRNA oligonucleotides (antagomiRs)62 or locked nucleic acid,63 miRNA sponges, erasers or masks. AntagomiRs are synthetic oligonucleotides with miRNAcomplementary sequences that bind to endogenous miRNAs competitively inhibit them to bind to their target genes. MiRNA sponges64,65 and erasers62 are sequences of multiple miRNA sequences incorporated into a vector (e.g. a [cardiotropic] virus). While sponges contain only the seed sequence and might therefore inhibit various miRNAs, erasers are complementary to specific miRNAs. MiR-masks, however, are single-stranded oligonucleotides that are complementary to a miRNA target sequence and can therefore specifically block single miRNA–mRNA interactions.35,66
All potential therapeutic interventions are currently based on an intramuscular or systemic application of these agents in vivo. Figure 1 summarises the suspected mechanisms of miRNAs being involved in AF pathophysiology and illustrates potential interventions in order to interrupt the vicious circle of AF begets AF.
Future Directions and Clinical Perspective
MiRNA manipulation in animal models has been demonstrated as a potential therapeutic approach. A first clinical trial used miravirsen, an antagomiR of miR-122, on patients with chronic hepatitis C infection (HCV).67 In this Phase IIa multicentre trial the use of miravirsen was shown to safe and effective in 36 patients: 5 weekly injections resulted in a dose-dependent reduction of HCV RNA levels for 14 weeks without evidence of viral resistance. A follow-up study on their patient cohort could confirm the safety and effectivity of miravirsen.68 Furthermore, upcoming clinical trials targeting miRNAs have been announced in the context of kidney fibrosis and coronary artery disease.69,70
These initial data and upcoming trials in humans are promising and justify further evaluation of miRNA therapeutics in AF, too. Based on the available data and suggested mechanisms of atrial fibrosis by miRNA action one focus could be the prevention or inhibition of progression of atrial fibrosis by targeting extracellular matrix-relevant miRNAs (e.g. miR-21 or miR-29b).
However, several challenges remain including more detailed evaluation of underlying pathophysiological mechanisms, chemical optimisation of miRNA agents or refinement of drug delivery. Furthermore, miRNAs might also serve as diagnostic biomarkers for AF as shown in our study on miR-29b. We demonstrated a significant downregulation of miR-29b in the plasma of patients with persistent AF that was further aggravated in patients with AF and concomitant congestive heart failure.54 In case miRNAs could provide a representative biomarker for the existing structural changes or the predominant underlying pathophysiological mechanism this information could guide the choice of therapy in the individual patient.
In summary, progress in miRNA research has opened a window for establishing a new potential therapeutic intervention in the context of translational medicine. The future will show whether miRNAs can help to close the translational gap between underlying causes and specific treatment, which is currently thought to be one major problem in AF disease management.71
Clinical Perspective
- MicroRNAs are mediators of electrical and structural remodelling leading to AF.
- MicroRNA-related mechanisms can be targeted in vivo by antagomiRs, locked nucleic acids, miRNA-ponges/
erasers, miR-masks (inhibiting miRNA effects) or miR mimics (enhancing miRNA effects). - A first clinical phase IIa trial using an antagomiR for treatment of hepatitis C demonstrated it to be safe and effective for treatment of human patients.
- Upcoming clinical trials will be targeting miRNAs in the context of kidney fibrosis and coronary artery disease.
- Targeting miRNAs in patients might be a novel therapeutic option for treatment of AF.