







The link between sudden unexplained death in individuals with mental health problems who are administered antipsychotic drugs has been recognised for over a century.1 A clear relationship has emerged over the past 25 years between antipsychotic drugs, prolongation of the QT interval of the ECG, atypical polymorphic tachycardia known as torsade de pointes (TdP) and sudden cardiac death (SCD). A number of antipsychotic drugs have been temporarily or permanently withdrawn from the market – or their use restricted – because of a concern over QT and QT corrected for heart rate (QTc) prolongation and development of TdP. In some cases, close follow-up with an ECG has been recommended or a modification of label imposed. The list of antipsychotic drugs implicated includes pimozide, sertindole, thioridazine, mesoridazine, promazine, triflupromazine, droperidol, moperone, pipamperone, sultopride and ziprasidone. A link between clinical use of antidepressants and the development of arrhythmias was first suggested following the Cardiac Arrhythmia Suppression Trial (CAST),2 based on the sodium channel-blocking properties of a number of antidepressant drugs including imipramine. Tricyclic antidepressants were subsequently shown to inhibit potassium channels and thus to prolong the QT interval and induce TdP. These effects of tricyclic antidepressants are generally observed when combined with other QT-prolonging agents or in cases of overdose. In addition, a number of case reports have linked tricyclic antidepressants as well as antipsychotic drugs to drug-induced Brugada syndrome (BrS), leading to syncope and SCD as a result of the development of rapid polymorphic ventricular tachycardia (VT) and VF.
Our focus in this article is on mechanisms and predisposing factors underlying the development of cardiac arrhythmias and SCD associated with antidepressant and antipsychotic drugs in clinical use.
Drug-induced Long QT Syndrome and Torsade de Pointes by Antidepressant and Antipsychotic Agents
The QT interval is a measure of the time interval between the start of depolarisation and the end of repolarisation. QTc intervals above 450 ms in men and 460 ms in women are considered to be abnormally prolonged. TdP, from the French for ‘twisting of the points’, is an atypical VT characterised by oscillations of the points or R wave peaks (‘pointes’) around the main axis of the ECG, giving rise to a unique morphology. Since the original work of François Dessertenne,3 it has been well recognised that many conditions are capable of causing prolonged or abnormal repolarisation, giving rise to QT prolongation, abnormal T/U wave morphologies and the development of TdP.4 Although a prolonged QT interval is essential for the development of TdP, it is generally not considered sufficient to induce TdP. An increased risk for TdP is recognised when the QTc exceeds 500 ms and whenever a drug increases QTc by >60–70 ms, especially when the increase develops rapidly.4–6 In addition to QTc prolongation, the risk of TdP is associated with the dispersion of transmural repolarisation and excitability. TdP arrhythmias can degenerate into VF, leading to SCD.
TdP can be caused by either congenital or acquired long QT syndrome (LQTS). Congenital LQTS is subdivided into 10 genotypes distinguished by mutations in at least 17 different genes encoding for cardiac ion channels and structural anchoring proteins. In the different genotypes, cardiac events may be precipitated by physical or emotional stress (LQT1), a startle (LQT2), or may occur at rest or during sleep (LQT3).
Acquired LQTS refers to a syndrome caused by circumstances other than genetic factors, including exposure to drugs that prolong the duration of the ventricular action potential (AP),7 or secondary to cardiomyopathies such as dilated or hypertrophic cardiomyopathy, as well as to abnormal QT prolongation associated with bradycardia or electrolyte imbalance.8–13 The acquired form of the disease is far more prevalent than the congenital form, and in some cases may have a genetic predisposition.
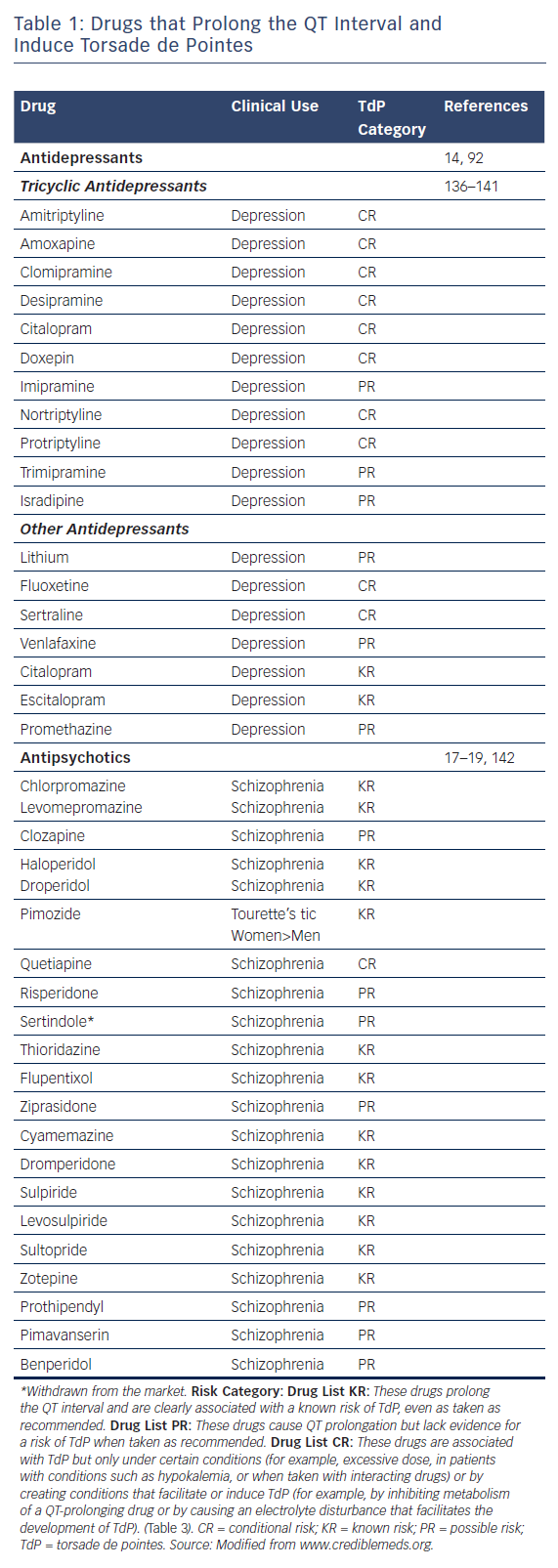
Table 1, modified from www.CredibleMeds.org, lists the antidepressant and antipsychotic drugs that have been shown to prolong QT interval and induce TdP. Among the antidepressant agents, amitriptyline, imipramine, and maprotiline are the agents most commonly associated with TdP the greatest prolongation of QT interval is observed with maprotiline).14 Table 1 shows the relative risk of TdP for distinct antidepressants and antipsychotics by classifying the drugs in TdP risk categories ranging from the highest risk of TdP; known risk of TdP (KR), to possible risk of TdP (PR) and conditional risk of TdP (CR). As shown in Table 1, most antidepressants are classified as CR, with a low risk of TdP. Vieweg and Wood14 reported 13 cases of TdP induced by antidepressants. As is typical of acquired LQTS, most cases (12 of 13) involved women. One case involved a child. In addition to female sex, risk factors include age (peaking in adolescence), bradycardia, metabolic inhibitors, hypokalemia, hypomagnesaemia, drug overdose and co-administration of QT-prolonging drugs (Table 2). QRS duration of the ECG, measured in five of 13 cases, showed prolongation in two and no change in three, suggesting that QT prolongation with antidepressants may be a result of sodium as well as potassium channel blockade. In a recent study, Danielsson and colleagues confirmed that antidepressants with KR or PR of TdP were associated with a higher risk than those classified as CR of TdP.15 Selective serotonin reuptake inhibitors (SSRIs), including citalopram, escitalopram, fluoxetine, paroxetine, sertraline and venlafaxine have been shown to exhibit a higher risk than tricyclic antidepressants, especially considering that an increase in dose of SSRIs, but not that of tricyclic antidepressants, markedly increases the risk of sudden death.16 For the most commonly used antidepressants in the elderly, the following risk ranking was observed, from highest to lowest; mirtazapine;citalopram;sertraline;amitriptyline.15 In 2011, the Food and Drug Administration (FDA) issued a warning concerning the antidepressant citalopram (Celexa®, Allergan) and its potential risk for TdP at doses greater than 40 mg (FDA, drug safety communication). Providers were asked to use doses less than 20 mg in patients over 60 years of age with hepatic dysfunction, as well as in poor CYP2C19 metabolisers or those taking concomitant CYP2C19 inhibitors. The maximum daily dosage of citalopram is now 40 mg in the non-elderly adult population. Most studies show that – in contrast to antipsychotics – antidepressants, including SSRIs, induce QT prolongation and TdP arrhythmias in cases of overdose, but rarely at therapeutic concentrations. However, QT prolongation may occur in elderly patients at therapeutic doses.16
Among the newer non-SSRIs, QT prolongation has rarely been reported with venlafaxine at therapeutic doses or with overdose. Bupropion has been linked to QT prolongation in overdose situations. In elderly patients with a number of high-risk comorbidities, mirtazapine demonstrated higher risk of SCD and ventricular arrhythmias than paroxetine.16 Jasiak et al. concluded that, based on the current literature, risk of QT/QTc prolongation with most newer non-SSRI antidepressants at therapeutic doses is low.16 The highest risk for QT prolongation appears to exist in overdose situations with venlafaxine and bupropion. They note that, given the few controlled studies and confounding variables in case reports, it is difficult to draw conclusions on QT prolongation risk with many of the newer non-SSRI antidepressants.
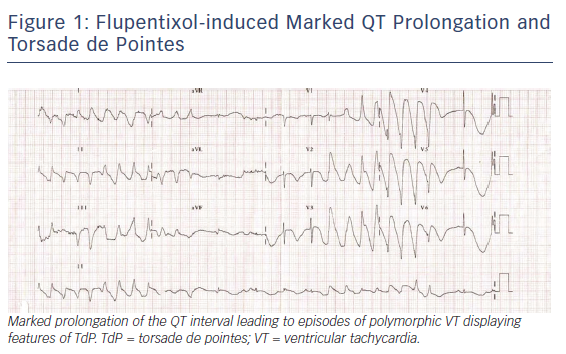
Antipsychotic drugs, especially those in the phenothiazine group, can also induce QT prolongation and TdP. Mehtonen et al. surveyed cases of sudden death by antidepressant or antipsychotic drugs and found 49 cases of sudden death (31 women and 18 men) associated with the use of these agents.17 A therapeutic dose of phenothiazine was involved in 46 of the 49 cases. Thioridazine was the only antipsychotic drug administered in 15 of the 49 cases. Figure 1 displays a case of a marked QT prolongation and TdP induced by the antipsychotic agent flupentixol.
Antipsychotic drugs generally have a higher torsadogenic potential (category KR and PR) than antidepressants (category CR) (Tables 1 and 3). As a consequence, antidepressant-induced TdP is more typically observed in the presence of drug combinations. Ray et al. conducted a retrospective cohort study of half a million Medicaid patients between 1988 and 1993, before the introduction of atypical antipsychotics, and observed that the risk for sudden death increased 2.39 times in individuals receiving antipsychotic drugs compared with those who did not receive these agents.18 Although the study did not demonstrate causality, it suggested that the potential adverse cardiac effects of antipsychotics should be considered in clinical practice, particularly for patients with cardiovascular disease. Hennessy et al. in a study of 90,000 patients, observed that those treated for schizophrenia had a higher incidence of cardiac arrest and ventricular arrhythmias than non-schizophrenia patients.19 The drugs used were clozapine, haloperidol, risperidone, and thioridazine. In a study of nursing home residents in six states, Liperoti et al. observed that the use of conventional antipsychotics led to a twofold increase in risk of hospitalisation for ventricular arrhythmias and cardiac arrest, especially in patients with pre-existing cardiac disease.20 In the elderly, haloperidol has been associated with high risk of TdP.15 The IV form of haloperidol is thought to carry a higher risk of QTc prolongation and TdP than the oral form.21,22 In 11 cases of fatal TdP, eight occurred with IV haloperidol. The FDA now recommends cardiac monitoring for all patients receiving haloperidol.
Drug-induced Brugada Syndrome Phenotype by Antidepressant and Antipsychotic Agents
BrS was introduced as a new clinical entity by Pedro and Josep Brugada in 1992.23 The syndrome has been associated with a high risk of sudden death, especially in men as they enter their third and fourth decades of life. A consensus report published in 2002 delineated diagnostic criteria for the syndrome.24,25 A second consensus conference report published in 2005 focused on risk stratification schemes and approaches to therapy.26,27 The most recent expert consensus report focused on emerging concepts, advances in risk stratification and approaches to therapy for both Brugada and early repolarisation syndrome (ERS), the so-called J wave syndromes.28
BrS is characterised by an electrocardiographic pattern of right bundle brunch in right precordial leads, ST-segment elevation in the right precordial leads, relatively normal QTc interval, coupled with syncope and sudden death caused by VT/VF in patients with no or minimal structural disease.
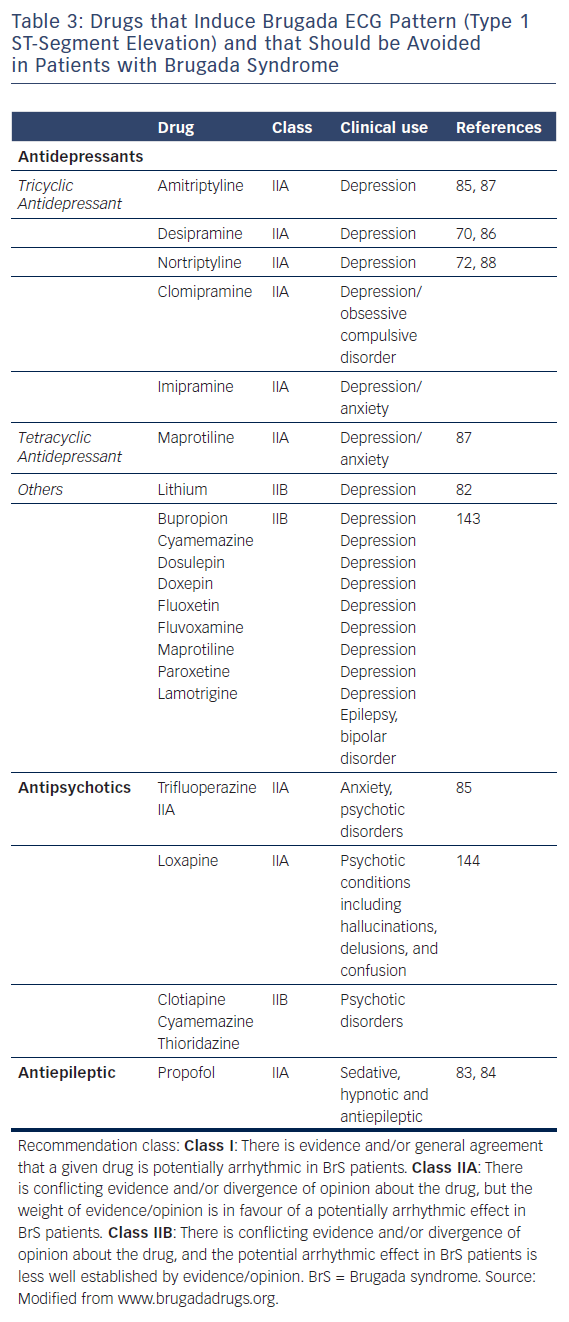
Three types of repolarisation patterns in the right precordial leads are recognised.24,25 Type 1 ST-segment elevation is diagnostic of BrS and is characterised by a coved ST-segment elevation ≥2 mm (0.2 mV) followed by a negative T wave. Type 2 ST-segment elevation has a saddleback appearance with a high take-off ST-segment elevation of ≥2 mm, followed by a trough displaying ≥1 mm ST elevation, followed by either a positive or biphasic T wave. Type 3 ST-segment elevation has either a saddleback or coved appearance with an ST-segment elevation of <1 mm. These three patterns may be observed sequentially in the same patient or following the introduction of specific drugs, particularly sodium channel blockers. Type 2 and type 3 ST-segment elevation are not considered to be diagnostic of BrS. BrS is definitively diagnosed only when a type 1 ST-segment elevation (Brugada ECG) is observed in more than one right-precordial lead (V1–V3), in the presence or absence of sodium channel-blocking agent, and in conjunction with one or more of the following; documented VF, polymorphic VT; a family history of SCD (<45 years old); coved type ECGs in family members; inducibility of VT with programmed electrical stimulation; syncope; or nocturnal agonal respiration.24–27
The average age at the time of cardiac arrest in patients with BrS is approximately 45 years, but most develop symptoms between 20 and 65 years.29–31 BrS in children is rare, but sudden death in this population is reported.32,33 Men are at increased risk for development of a spontaneous type I Brugada ECG and SCD.34,35
In a recent study, women comprised 42 % of a cohort of 542 patients who presented with a spontaneous or drug-induced Brugada type 1 pattern on the ECG.36 Women with BrS present more benign clinical characteristics, less spontaneous type 1 ECG pattern, and are more likely to be asymptomatic than men.35
BrS has been associated with variants in 19 different genes. The gene most often associated with BrS is SCN5A, accounting for 11–28 % of cases, depending largely on geographic location. Over 300 BrS-related variants in SCN5A, the gene encoding the alpha subunit of the cardiac sodium channel, have been reported.37–40 Loss-of-function mutations in SCN5A contribute to the development of both BrS and ERS, as well as to various conduction diseases, Lenegre’s disease and sick sinus syndrome.
Variants in genes encoding the calcium channels including CACNA1C (Cav1.2), CACNB2b (Cavbeta2b) and CACNA2D1 (Cavalpha2delta) have been reported in up to 13 % of probands.41–44 Mutations in glycerol-3-phosphate dehydrogenase 1-like enzyme gene (GPD1L), SCN1B (beta1-subunit of sodium channel), KCNE3 (MiRP2), SCN3B (beta3-subunit of soduim channel), KCNJ8 (Kir6.1), KCND3 (Kv4.3), RANGRF (MOG1), SLMAP, ABCC9 (SUR2A), (Navbeta2), PKP2 (plakophillin-2), FGF12 (FHAF1), HEY2, SEMA3A (semaphorin) and KCNAB2 (Kvbeta2) are relatively rare.45–56 An association of BrS with SCN10A, a gene encoding a neuronal sodium channel, was first reported in 2014.56–58 There is controversy as to the pathogenicity of many SCN10A mutations with yields ranging from 5.0 % to 16.7 %.57–59 Mutations in all of these genes lead to loss of function in sodium (INa) and calcium (ICa) channel currents, as well as to a gain of function in transient outward potassium current (Ito) or ATP-sensitive potassium current (IK-ATP).58,60
New susceptibility genes proposed and awaiting confirmation include the transient receptor potential melastatin protein 4 gene (TRPM4)61 and the KCND2 gene. Variants in KCNH2, KCNE5, SEMA3A, although not causative, have been identified as capable of modulating the substrate for the development of BrS.62–65 KCNE4 has recently been added to this group (unpublished observation, Clatot and Antzelevitch). Loss-of-function mutations in HCN4 (prominently expressed in the sinus node) have been associated with BrS but may be modulatory by acting to unmask BrS by reducing heart rate.66
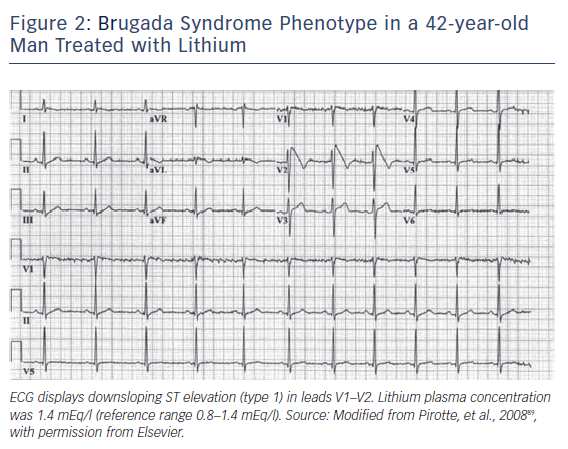
A large number of factors modulate the electrocardiographic and arrhythmic manifestations of BrS. ST-segment elevation in BrS is often dynamic. The Brugada ECG may be concealed, but can be unmasked or modulated by sodium channel blockers, a febrile state, vagotonic agents, alpha adrenergic agonists, beta adrenergic blockers, tricyclic or tetracyclic antidepressants, first generation antihistamines (dimenhydrinate), a combination of glucose and insulin, hyperkalemia, hypokalemia, hypercalcaemia, and by alcohol and cocaine toxicity.67–77 These agents may also induce acquired forms of BrS. Propafenone, typically prescribed for the treatment of AF, is a common example of a drug that can unmask BrS.78–81 Lithium, a widely used antidepressant agent, has been recently added to the list of drugs to avoid in patients with BrS. Lithium is a potent blocker of cardiac sodium channels and can unmask a type 1 ECG in patients with BrS.82 Propofol, a short-acting, IV-administered sedative-hypnotic and antiepileptic agent with anaesthetic properties, may cause a rare condition called propofol infusion syndrome, characterised by unexplained lactic acidosis, lipaemia, rhabdomyolysis, cardiovascular collapse and Brugada-like ECG pattern following high-dose propofol infusion over prolonged periods of time.83,84
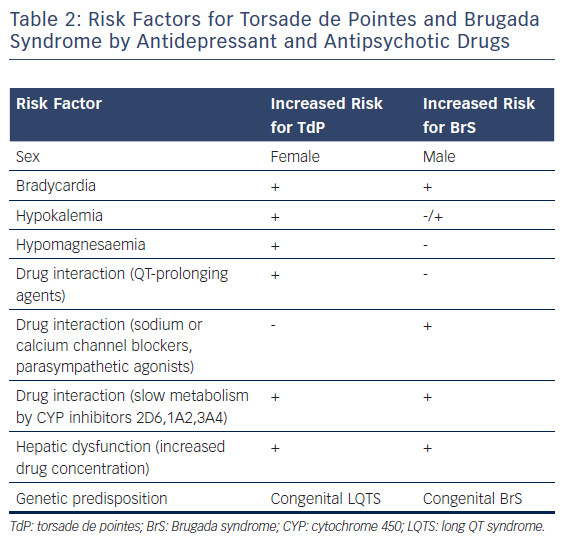
Table 3 lists the antidepressants and antipsychotic agents reported to induce the Brugada ECG pattern. All cases of BrS pattern induced by antidepressants and antipsychotics displayed a type 1 ST-segment elevation (Figure 2). A study of 98 patients experiencing an overdose of tricyclic antidepressants reported that 15 of these displayed an ECG consistent with BrS.71 The overall mortality was 3.0 % among all patients, but 6.7 % among patients who displayed a Brugada phenotype. Rouleau et al. described three cases of psychotropic drug-induced Brugada ECG,85 occurring during concomitant administration of amitriptyline and a phenothiazine (case 1), overdose of fluoxetine (case 2), and co-administration of trifluoperazine and loxapine (case 3). Babaliaros and Hurst described a Brugada pattern in patients receiving increasing doses of imipramine.70 Akhtar and Goldschlager reported a case of BrS following massive ingestion of desipramine and clonazepam.77 Chow et al. reported a similar case involving desipramine.86 Bolognesi et al. described a Brugada ECG pattern following overdose of amitriptyline and with maprotiline.87 Additional cases of BrS were reported following overdose with nortriptyline72,88 or lithium.82,89
The available data suggest that most cases of antidepressant and antipsychotic-induced BrS phenotype occur as a consequence of drug overdose or drug combination.
Effects of antidepressants and antipsychotics on ion channels
Antidepressants and antipsychotics are reported to modulate the cardiac AP by blocking a variety of cardiac ion channels. In the ventricle they inhibit the fast sodium channel inward current (INa), the inward calcium current (ICa), and one or more outward potassium currents (IK), particularly the rapidly activating delayed rectifier current (IKr). Drug-induced IKr block has attracted considerable attention in recent years because of the association of IKr block with QT interval prolongation in the ECG and life-threatening cardiac arrhythmias such as TdP. Drug-induced INa and ICa block underlie the development of the BrS phenotype in experimental models of BrS.90,91
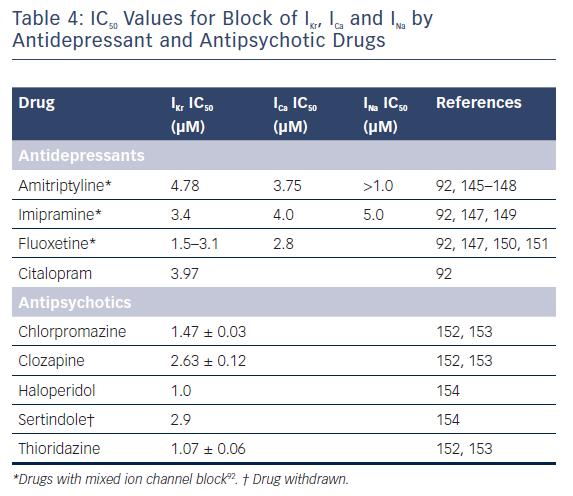
Table 4 illustrates the IC50 values for block of IKr, ICa and INa derived from heterologous expression systems (for example, HEK and CHO cells) and/or native cardiac myocytes for a number of antidepressants and antipsychotics that have been shown to induce arrhythmias.92 The available studies suggest that most antidepressants inhibit both inward and outward currents, these include imipramine, amitriptyline, and fluoxetine, all of which block both IKr and ICa. Imipramine and amitriptyline also block INa. The ability of antidepressants to block both outward and inward currents is associated with lack of correlation between the degree of IKr block and QT prolongation because calcium and/or sodium channel inhibition limit the effects of IKr block to prolong AP duration (APD) and thus to prolong the QT interval. In contrast to antidepressants, antipsychotic drugs produce more of an outward current inhibition. QT prolongation is most commonly secondary to inhibition of IKr. A 30-fold difference between the effective plasma concentration and the IC50 for inhibition of IKr has been suggested as an adequate margin of safety for avoiding the development of TdP as an adverse effect.93
Mechanisms of Arrhythmias in Long QT Syndrome
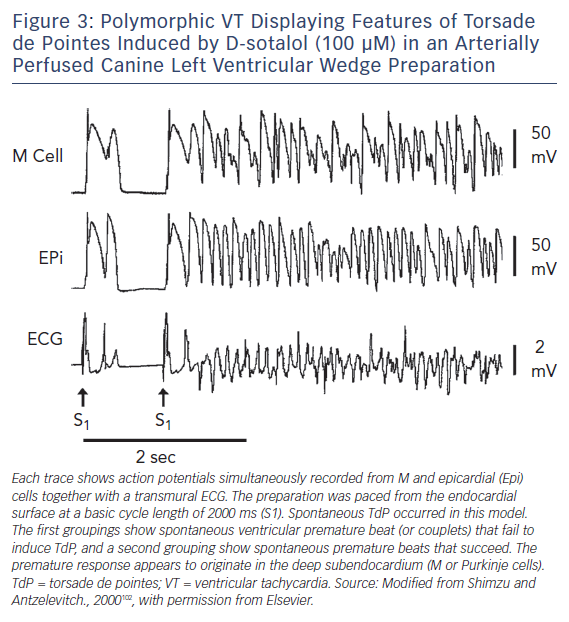
Amplification of spatial dispersion of repolarisation within the ventricular myocardium has been identified as the principal arrhythmogenic substrate in both acquired and congenital LQTS. The accentuation of spatial dispersion – typically secondary to an increase of transmural, trans-septal or apico-basal dispersion of repolarisation – and the development of early afterdepolarisation (EAD)-induced triggered activity, underlie the substrate and trigger, respectively, for the development of TdP arrhythmias observed under LQTS conditions.94,95 Models of the LQT1, LQT2, LQT3, LQT5, LQT6, LQT7, and LQT8 forms of the LQTS have been developed using the canine arterially-perfused left ventricular wedge preparations.96–99 These models suggest that in the first three forms of LQTS preferential prolongation of the M cell APD leads to an increase in the QT interval, as well as an increase in transmural dispersion of repolarisation (TDR), which contributes to the development of TdP (Figure 3).100–102 The unique characteristics of the M cells are at the heart of the LQTS. The hallmark of the M cell is the ability of its AP to prolong more than that of endocardium or epicardium in response to a slowing of rate.103–105 This feature of the M cell is a result of weaker repolarising current during phases two and three of the AP, secondary to a smaller IKs and a larger late INa and INa-Ca.106–108
These ionic distinctions also sensitise the M cells to a variety of pharmacological agents. Agents that block IKr (such as antidepressants and antipsychotics), IKs, or increase ICa or late INa, generally produce a much greater prolongation of the APD of the M cell than that of epicardial or endocardial cells. The duration of the M cell AP therefore determines the QT interval, whereas the duration of the epicardial AP generally determines the QT peak interval.
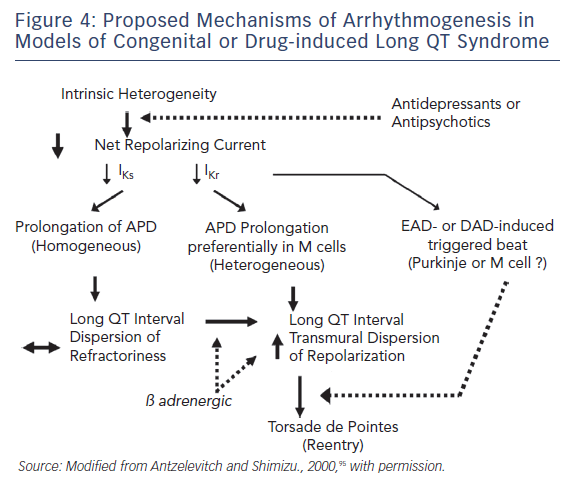
Figure 4 presents our working hypothesis of the mechanisms underlying LQTS-related TdP. The hypothesis presumes the presence of electrical heterogeneity in the form of spatial dispersion of repolarisation in the form of transmural and trans-septal dispersion of repolarisation under baseline conditions, and the amplification of TDR by agents that reduce net repolarising current via a reduction in IKr or IKs (or augmentation of ICa or late INa). Pharmacological agents or other conditions that cause a reduction in IKr lead to a preferential prolongation of the M cell AP. As a consequence, the QT interval prolongs and is accompanied by a dramatic increase in TDR, creating a vulnerable window for the development of re-entry. The reduction in net repolarising current also predisposes to the development of EAD-induced triggered activity and, in rare cases, delayed afterdepolarisation-induced, triggered activity in M and Purkinje cells, which provide the extrasystole that triggers TdP when it falls within the vulnerable period. Betaadrenergic agonists further amplify transmural heterogeneity in the case of IKs block, as well as (transiently) in the case of IKr block, but reduce it in the case of INa agonists.102,109 Inhibition of IKr is the most common cause of reduction in net outward current by antidepressant and antipsychotic drugs. The presence of other IKr blockers (combination of an antidepressant and antipsychotic drug) or agents that reduce IKs or augment ICa or late INa can accentuate the reduction in repolarisation forces and increase the probability of arrhythmia.
Mechanisms of Arrhythmia in Brugada Syndrome
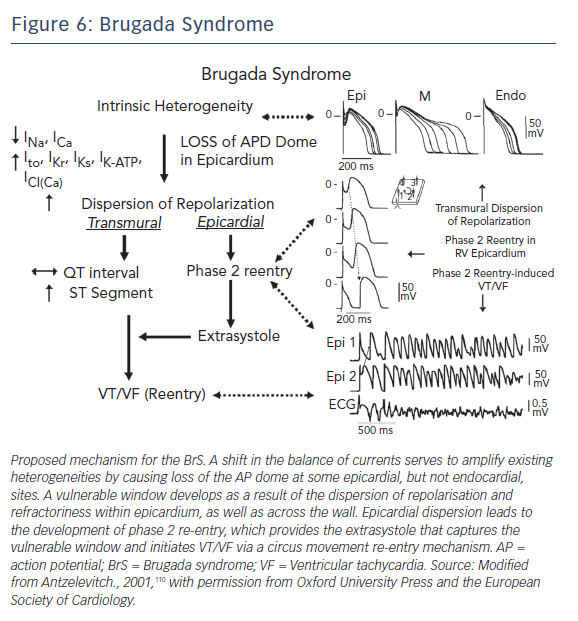
The development of prominent J waves, appearing as ST-segment elevation in the right precordial leads of BrS patients, is believed to be a result of accentuation of the right ventricular (RV) epicardial AP notch secondary to an outward shift in the balance of currents active at the end of phase 1.110 A spike and dome morphology, displaying a prominent notch in ventricular epicardium but not endocardium, generates a transmural voltage gradient that results in the electrocardiographic J wave.111
The cellular basis for BrS is thought to involve an outward shift of net transmembrane current active at the end of phase 1 of the RV epicardial AP. The Ito has been shown to be most prominent in the right ventricle, particularly in the region of the RV outflow tract (RVOT).112 Such a shift can accentuate the AP notch and lead to all-or-none repolarisation at the end of phase 1 (Figures 5 and 6). When phase 1 repolarises beyond the voltage range at which L-type Ca+2 channels activate, the Ca+2 channels fail to activate, resulting in loss of the AP dome. Conduction of the AP dome from epicardial sites at which it is maintained to sites at which it is lost gives rise to phase 2 re-entry that generates a closely coupled extrasystole that precipitates VT/VF.110,113,114
Although genetic mutations are equally distributed between sexes, the clinical phenotype is 8– to10–times more prevalent in men than in women. The basis for this sex-related distinction is a more prominent Ito in the RV epicardium of men versus women.115 The more prominent Ito-mediated AP notch causes the end of phase 1 of the RV epicardial AP to repolarise to more negative potentials in tissue and arterially-perfused wedge preparations from men, facilitating loss of the AP plateau and the development of phase 2 re-entry and polymorphic VT.
The cellular mechanisms underlying BrS have long been a matter of debate.116,117 Two principal hypotheses have been proposed; the repolarisation hypothesis and the depolarisation hypothesis. The repolarisation hypothesis described above maintains that an outward shift in the balance of currents in RV epicardium can lead to repolarisation abnormalities, resulting in the development of the substrate for re-entrant activity as well as the development of phase 2 re-entry, which generates closely coupled premature beats capable of precipitating VT/VF. The depolarisation hypothesis suggests that slow conduction in the RVOT, as a result of fibrosis, reduces Cx43 expression leading to discontinuities in conduction. Conduction slowing is not necessarily limited to the RVOT area.
Leong et al. recently reported a study in which the magnitude of ST elevation correlated with the degree of ajmaline-induced conduction delay in the RVOT of patients with type I Brugada ECG, seemingly supporting the depolarisation hypothesis.118 The study included 11 patients with concealed type I BrS ECG and two healthy controls undergoing ECG imaging before and after ajmaline infusion. Activation maps and activation recovery intervals were derived from electrograms recorded from the epicardial surface of the heart, including the RV, RVOT, and left ventricle (LV). Conduction time was recorded from 3.5 cm segments within these regions of the heart before and after ajmaline and correlated with J point (ST-segment) elevation observed in the surface ECGs. Ajmaline increased conduction delay by 5.4 ± 2.8 ms in the RVOT, 2.0 ± 2.8 ms in RV free wall and 1.1 ± 1.6 ms in LV free wall. Conduction delay in the RVOT, but not RV or LV, correlated with the degree of J point elevation in BrS patients. The authors’ conclusion that the magnitude of J point (ST-segment) elevation in patients with the type I BrS pattern is attributable to conduction delay in the RVOT was challenged by Antzelevitch and Patocskai who demonstrated that ajmaline can induce prominent ST-segment elevation by accentuation of the RV epicardial AP notch.119 They further pointed out that according to the depolarisation hypothesis for ST-segment elevation associated with BrS, RVOT activation delay, relative to activation of the RV, must be roughly equivalent to the duration of the ST segment elevation (typically >200 ms)116 and that the 3.4 ms reported by Leong et al. falls far short of that requirement, thus discounting the depolarisation hypothesis as a cause.
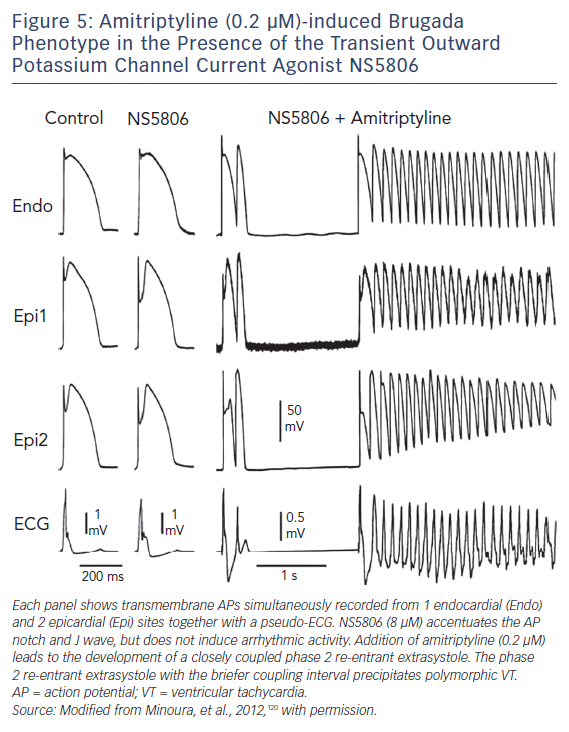
Figure 5 displays an example of amitriptyline-induced BrS phenotype in the presence of the Ito agonist NS5806 in a canine right ventricular wedge model.120 Amitriptyline (0.2 μm) caused loss of the AP dome in the AP of Epi1 but not in that of Epi2. Phase 2 re-entry developed as the epicardial AP dome propagated from sites at which it was maintained to sites at which it was lost. This mechanism generates closely coupled extrasystoles and the development of a polymorphic VT.
Genetic Predisposition
The degree to which a genetic predisposition contributes to the clinical manifestation of antidepressant- and antipsychotic-induced arrhythmogenesis is not well defined.
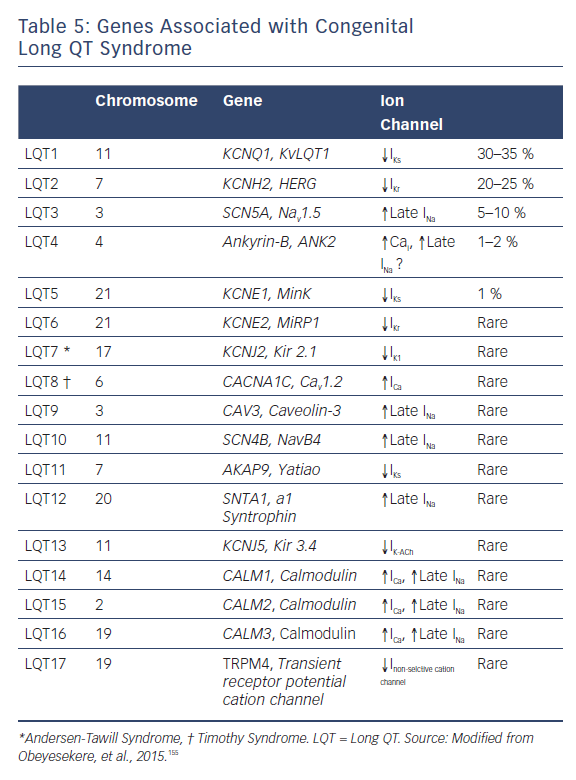
Congenital LQTS has been associated with 17 different genes (Table 5). Drugs are by far the most common cause of acquired forms, including drug-induced forms, of LQTS. Mounting evidence suggests that drug-induced LQTS also has a significant heritable component, and recent studies have made advances in identifying the genetic substrate underlying drug-induced LQTS. Advances in next-generation sequencing technology and molecular biology techniques have identified genetic variants underlying the acquired form of LQTS.121
Acquired LQTS characterised by QT prolongation and TdP triggered by drugs, hypokalemia or bradycardia are usually reversed upon elimination of the triggers. In some cases the LQTS phenotype persists, suggesting the presence of an underlying genetic substrate.
Itoh et al. reported that a third of acquired LQTS patients carry congenital LQTS mutations, with variants in KCNH2 being the most common.122 In a recent study, Strauss et al. demonstrated that a genetic QT score comprising 61 common genetic variants can account for a significant proportion of the variability in drug-induced QT prolongation and is a significant predictor of drug-induced TdP.123 The authors indicate that these findings highlight an opportunity for such genetic discoveries to improve individualised risk–benefit assessment for pharmacologic therapies. Of note, replication of these findings in larger samples is needed to more precisely identify the individual variants that drive these effects.123
Abbott et al. were among the first to show that a polymorphism (a genetic variation that is present in greater than 1 % of the population) in an ion channel gene is associated with a predisposition to drug-induced TdP.124 They identified a polymorphism (T8A) of the KCNE2 gene encoding for MiRP, a beta subunit of the IKr channel, that is present in 1.6 % of the population and is associated with TdP related to quinidine and to sulfamethoxazole/trimethoprim administration. This finding suggests that common genetic variations may increase the risk for development of drug-related arrhythmias. Yang et al. showed that DNA variants in the coding regions of congenital long QT disease genes predisposing to acquired LQTS can be identified in approximately 10–15 % of affected subjects, predominantly in genes encoding ancillary subunits, providing further support for the hypothesis that subclinical mutations and polymorphisms may predispose to drug-induced TdP.125 Splawski et al. further advanced this concept by identifying a heterozygous polymorphism involving substitution of serine with tyrosine in codon 1103 (S1103Y) in the sodium channel gene SCN5A (S1102Y in the shorter splice variant of SCN5A) among Africans and African-Americans that increases the risk for acquired TdP.126 The polymorphism was present in 57 % of 23 patients with pro-arrhythmic episodes, but in only 13 % of controls.
Another common polymorphism that has been associated with acquired forms of LQTS and TdP is K897T in KCNH2.127 Most functional expression studies have reported that K897T reduces IKr,128–130 although one study has reported an increase in hERG current.131
Antidepressant drugs have been shown to induce acquired LQTS via both direct inhibition of the hERG or IKr channel or via impaired trafficking of the channel to the surface membrane, thus mimicking the effects of some KCNH2 variants.132
The action of antidepressants to precipitate the BrS may also have a genetic disposition. For example, the SCN5A promoter haplotype (so-called Hap B) has been shown to be associated with longer PR and QRS intervals as well as with a more exaggerated response to sodium channel blockers.133
Genetic defects can also contribute to drug-induced channelopathies by influencing the metabolism of drugs. In the case of relatively pure IKr blockers, there is a clear relationship between plasma levels of drug and the incidence of TdP. Genetic variants of the genes encoding for enzymes responsible for drug metabolism could alter pharmacokinetics so as to cause wide fluctuations in plasma levels, thus exerting a significant proarrhythmic influence.134,135 For example, in the case of cytochrome CYP2D6, which is involved in the metabolism of some QT-prolonging drugs (terodiline, thioridazine), multiple polymorphisms have been reported that reduce or eliminate its function; 5–10 % of Caucasians and African-Americans lack a functional CYP2D6. Numerous proteins, including drug transport molecules and other drug metabolising enzymes, are involved in drug absorption, distribution and elimination, and genetic variants of each of these has the potential to modulate drug concentrations and effects. Multiple substrates and inhibitors of the cytochrome P450 enzymes have been identified. A comprehensive database can be found at http://medicine.iupui.edu/flockhart
Antipsychotic drugs are more commonly associated with QT prolongation and TdP than antidepressants are. Most cases of antidepressant-induced TdP occur following drug overdose or when administered in combination with other QT-prolonging agents or conditions. Antidepressants, on the other hand, are more likely to predispose to BrS phenotype. These proclivities are because antipsychotic drugs generally exert a predominant effect to inhibit outward currents, IKr block in particular, whereas antidepressants exert a predominant effect to inhibit inward currents, such as INa and ICa. Commonly used antipsychotic and antidepressant drugs should be used with great care in cases of long QT or BrS,or when combined with agents known to prolong QT intervals or to predispose to acquired forms of BrS.
Acknowledgements
The authors acknowledge Gan-Xin Yan, Lin Wu and Subramanian Krishnan’s assistance in providing us with ECGs of patients administered with antipsychotic and antidepressant drugs.
Clinical Perspective
- Some antipsychotic and antidepressant drugs increase the risk of ventricular arrhythmias and SCD by prolonging the QT interval and inducing Torsade de Pointes arrhythmias. These include typical antipsychotics such as chlorpromazine; tricyclic antidepressants such as amitriptyline and other antidepressants such as fluoxetine.
- Other antipsychotic and antidepressant drugs increase the risk of ventricular arrhythmias and SCD by inducing a Brugada Syndrome phenotype. These include antipsychotics such as trifluoperazine; tricyclic antidepressants such as amitriptyline or desipramine; and other antidepressants such as maprotiline or lithium.
- Antipsychotic drugs can increase cardiac risk even at low doses, whereas antidepressant drugs generally do it at high doses or in combination with other drugs.
- The newer atypical antipsychotics, including olanzapine, risperidone, quetiapine, prothipendyl, pimavanserin and benperidol display a lower level of risk than the older typical antipsychotics, especially those in the phenothiazine category.
- Antipsychotic and antidepressant drugs should be used with great care in cases of long QT or BrS or when combined with agents known to prolong QT intervals or to predispose to acquired forms of BrS.
- In the case of drugs categorised as having a potential to cause significant QT prolongation and/or TdP, ECG monitoring is advisable, particularly where the FDA-approved label recommends ECG monitoring. Review of specific antipsychotic or antidepressant therapy, including cessation and change of medication should be considered if the ECG shows major prolongation of the QT interval (QTc >500 ms), QTc prolongation >60 ms, T wave abnormalities, marked bradycardia, or a BrS phenotype.
- Finally, high-risk antipsychotics and antidepressants should be avoided in patients with acute systemic disease, including acute MI and renal or hepatic disease.