







Hypertrophic cardiomyopathy (HCM), a genetic sarcomeric disorder associated with myocyte disarray and scar deposition, is intimately linked to sudden cardiac death (SCD) due to malignant ventricular arrhythmias. In the first modern published description of the disease in 1958, Dr Donald Teare describes the case of a 14-year-old male who collapsed while being chased around his school’s playground.1 He was reported to have been having ‘blackout episodes’ for months. He had seen physicians and the only abnormal finding elicited was a third heart sound and a soft systolic murmur. As there was no evidence of seizure activity, he was advised to continue his daily activities without restriction. After collapsing while playing, he was pronounced dead on arrival at the hospital and his autopsy was remarkable for massive septal hypertrophy. Dr Teare’s report described seven more cases of young adults all who suddenly collapsed without warning with similar findings on autopsy.
Since 1958 our understanding of HCM has markedly improved, but up to the 1980s little progress had been made in preventing SCD. The advent of the implantable cardioverter-defibrillator (ICD) has drastically changed our ability to prevent SCD. In this review we discuss our current epidemiological, genetic and structural understanding of the link between HCM and SCD. We also discuss the role of modern ICD therapy in the primary prevention of sudden death in patients with HCM.
Epidemiology of Hypertrophic Cardiomyopathy and Sudden Cardiac Death
HCM is the most common form of genetically inherited cardiovascular disease, with a prevalence of one in 500 individuals.2,3 Patients with HCM make up 1 % of cardiology practice. Many patients with HCM interact with the healthcare system from young adulthood onwards.4 The mortality rates in early studies were as high as 5–7 % per year.5,6 With the advent of ICDs and early detection of the disease, modern cohorts have mortality rates of under 1 %.7,8 SCD due to sustained ventricular tachycardia (VT) or ventricular fibrillation (VF) is still the most common cause of mortality, however, accounting for 51 % of all HCM-related deaths.8 Sudden death in patients with HCM is one of the leading causes of death in individuals under the age of 40.9 While there appears to be no difference in SCD rates based on gender, age is an important factor:10 SCD is more common in younger patients, especially those under the age of 35; however, up to 20 % of SCDs have been reported to occur in patients over the age of 65.8 Identifying patients within this heterogeneous disorder who are at high risk of sudden death is a challenge.
Genetics of Hypertrophic Cardiomyopathy and Sudden Cardiac Death
HCM is a heterogeneous disease that is classically transmitted in an autosomal dominant fashion. HCM often arises from mutations in genes responsible for the formation of cardiac sarcomeres. Sarcomeric mutations, initially discovered in the 1980s, are found in up to 70 % of patients with a family history of HCM.11 The most common mutations are in the myosin heavy chain (MYH7 gene) and myosin binding protein C (MYBPC3 gene).12 There is marked heterogeneity in penetrance, however, and a poor understanding of how the different genotypes manifest as HCM phenotypes. Currently, there is minimal evidence to support the use of genetic testing to identify patients who are at high risk of SCD. Small studies have found that patients with a more malignant inheritance of their mutations (i.e. homozygous mutations) as well as the presence of a MYH7 gene mutation have increased rates of heart failure.13 Emerging data suggest that rarer forms of the MYH7 mutation increase the risk of SCD.13,14 At this time, however, routine genetic testing cannot reliably be used to identify patients with HCM who are at elevated risk of SCD.
Structural Predisposition Towards Sudden Cardiac Death
Myocardial fibrosis, microvascular ischaemia and cellular disarray predispose patients with HCM to re-entrant ventricular arrhythmias. Histological findings in HCM are consistent with myocardial fibrosis due to scarring and disruption of the normal cellular architecture.15,16 Fibrosed and disrupted cellular architecture is often found throughout the heart, but is mostly concentrated in the densest area of hypertrophy.17 Silent low-grade myocardial ischaemia and altered coronary blood flow are common in HCM and help create the conditions needed for scar formation and arrhythmogenesis.18
Fibrosis can be evaluated noninvasively with advanced imaging techniques. Currently cardiac magnetic resonance imaging (cMRI) is being used to better understand the role of fibrosis and how it relates to disease progression and risk of SCD. Late gadolinium contrast enhancement (LGE) visualised on cMRI is being studied as a surrogate for the degree and distribution of fibrosis. An association between the presence of LGE and nonsustained ventricular tachycardia (NSVT) has been recorded by outpatient ambulatory monitoring.19 While no clear link between LGE and SCD has been established, a recent metaanalysis found that the risk of SCD increased in the presence of LGE (odds ratio (OR) 2.52; 95 % confidence interval (CI) [1.44–4.40]).20
Primary Prevention of Sudden Cardiac Death
While uncommon, SCD is the most devastating consequence of HCM. Prior to the advent of ICD therapy, pharmacological therapy was used in an attempt to reduce the risk of sudden death. The initial strategies involved beta-blockers and calcium-channel blockers, as well as antiarrhythmic agents such as quinidine, procainamide, sotalol and amiodarone. In a retrospective study prior to the widespread use of ICDs, the authors studied 293 patients who were considered to be at high risk for SCD.21 They compared the risk of SCD between patients who were prescribed medications (beta-blockers, verapamil, sotalol and amiodarone) for the prevention of SCD or relief of HCM symptoms versus those on no medical therapy. Patients on medical therapy had similar rates of sudden death to those receiving medications.21 It appears that pharmacological therapy alone is not sufficient to prevent SCD. In the current era of ICD implantation there are many patients on antiarrhythmic therapy who still receive appropriate ICD discharges to terminate VT/VF.22 Current and previous guidelines have found that there is no evidence for the use of medical therapy alone in the prevention of SCD.23,24
The advent of the transvenous ICD has markedly changed our ability to prevent SCD. It is of historical interest that two of the first individuals who had an ICD implanted were patients with HCM and frequent ventricular arrhythmias.25 The efficacy of ICD implantation specifically for the prevention of SCD in the HCM population was first studied in a group of 128 high-risk patients. In these patients, the rate of ICD therapy to terminate VT or VF was 7 % per year.22 Since then, several large multicentre studies have been completed that support the findings that ICDs are effective at terminating malignant arrhythmias with rates of therapy between 4 and 6 % per year.26–29 A multicentre international ICD registry consisting of 500 patients with HCM and ICD implants revealed that ICD interventions successfully terminated lifethreatening arrhythmias in 103 out of 104 patients who developed VT or VF.26 The one patient who was not successfully treated was found to have a faulty device. Another modern cohort study followed 1,000 patients under the age of 60 for 20 years.8 The authors found that the overall mortality rate for patients with HCM was similar to that of the general population, at nearly 1 %. While the mortality rates were similar, the cause of death was very different in both groups, with SCD being the major cause of mortality in the HCM group. Eighty per cent of the patients who suffered SCD had either declined ICD implantation or had been evaluated prior to the widespread use of ICD therapy. It is postulated that nine ICDs need to be implanted to treat one episode of malignant arrhythmia or SCD. This is comparable to high-risk patients with an ischaemic cardiomyopathy.30 There is no doubt that the ICD is efficacious in preventing SCD. The primary difficulty for the clinician arises when trying to select patients for whom the risks are outweighed by the benefits of ICD implantation.
Risk Stratification
Although the current mortality rate of HCM is low, there are groups of patients with an elevated risk of SCD who would benefit from ICD placement. The current American College of Cardiology Foundation (ACCF)/American Heart Association (AHA) guidelines from 2011 focus on the clinical factors that increase the risk of SCD.24 It is important to note that our current understanding is still incomplete. As always, one must tailor the decision to the needs and desires of the individual patient.
The guidelines recommend that all patients should undergo SCD risk stratification at their initial evaluation, as well as periodic re-evaluation to determine whether their risk of SCD has changed.24 The clinical variables are grouped into two categories (see Table 1). The first category includes established risk markers. Some of these markers are individually sufficient to prompt ICD placement, while others prompt ICD placement when they are found in conjunction with other high-risk features. The second category includes risk modifiers. These serve as additional markers to help identify high-risk patients; alone they are not sufficient to promote ICD placement. Established risk markers carry more weight, as the evidence linking them to SCD is more robust; however, our understanding of risk modifiers has continued to develop since the guidelines were produced in 2011.
Established Risk Markers
Prior Personal History of Ventricular Fibrillation, Sudden Cardiac Death or Sustained Ventricular Tachycardia
Patients who have already experienced and survived an episode of SCD or malignant ventricular arrhythmia represent the highest risk group. The annual rate of subsequent events in this secondary prevention group is 10 % per year.31–33 This is the only risk marker with an excellent positive predictive value.
Family History of Sudden Cardiac Death
Prior to our understanding that HCM is a genetic disease, authors made the observation that SCD disproportionately affects certain families.34 The first patient clinically diagnosed with HCM came to medical attention due to several siblings dying suddenly at a young age.35 Despite this, the lack of clear associated genetic markers has led some authors to argue that a family history should not be an established risk factor.36 Another complicating factor is the various definitions of family history used by investigators. Some studies use an age cut-off of 50 years when determining whether a SCD event was due to HCM, while others consider having had two first-degree relatives die from SCD as an indication of a positive family history.31,37 Recent studies define a positive family history as SCD in any first-degree relative of a patient with HCM.38 The totality of evidence appears to favour using a carefully elucidated family history as a risk factor for SCD. Cohort studies demonstrate that family history is independently associated with a 20 % increase in the relative risk of SCD.35 The ACCF/AHA guidelines define family history as an established risk marker if one or more first-degree family members have suffered SCD, irrespective of the family member’s age or whether he or she had a documented history of HCM.24
Unexplained Syncope
Syncope in HCM can originate from either a haemodynamic- or arrhythmia-mediated cause. Haemodynamic mechanisms include left ventricular outflow tract (LVOT) obstruction and abnormal vagal tone. A large series of 1,511 patients found that a history of unexplained syncope occurring 6 months prior to clinical evaluation led to a fivefold increase in the relative risk of SCD (adjusted hazard ratio: 4.89; 95 % CI [2.19–10.94]).39 A remote history of unexplained syncope (greater than 5 years prior to evaluation), however, did not increase the risk of SCD.39 The current guidelines consider any unexplained recent syncopal event identified after a careful history-taking to be a risk factor, and one that has occurred within the past 6 months to be particularly concerning.
Maximal Left Ventricular-Wall Thickness
There is a linear relationship between left ventricular hypertrophy (LVH) and the risk of SCD. An increase in LV mass leads to myocardial remodelling and fibrosis, which predispose patients to re-entrant arrhythmias.40 Studies have demonstrated that a LV-wall thickness ≥30 mm is an independent risk factor for SCD, with a 20 % increase in the relative risk of death at 10 years compared to the general HCM population.41 Based on these studies, the guidelines recommend a LV-wall diameter ≥30 mm be considered an established risk marker. It is important to note that LVH appears to increase the risk of SCD in younger patients to a greater extent. The guidelines recommend individualised decision-making when a younger patient begins to demonstrate LVH.41 Young patients (<30 years), even those with moderate LVH (LV-wall thickness >16 mm), should undergo further risk stratification.42 If one identifies other high-risk features such as NSVT, abnormal blood pressure response to exercise or fibrosis on MRI, then ICD implantation should be considered.
Nonsustained Ventricular Tachycardia
NSVT is defined as three or more consecutive ventricular beats of <30 s in duration at a heart rate ≥120 beats/min. It is most often found during ambulatory monitoring. Early studies failed to show NSVT as a clear, independently-associated risk factor for SCD.43However, a much larger contemporary study of 531 patients demonstrated that those who developed NSVT of any duration were more likely to die from SCD, especially younger patients. The OR for patients under the age of 30 with NSVT was 4.35 (95 % CI [1.54–12.28]); the OR for those >30 years was 2.16 (96 % CI [0.82–5.69]).44 There are conflicting reports on whether the duration and frequency of NSVT have any prognostic implications. The current ACCF/AHA guidelines suggest that there may be value in longer-term monitoring to help assess the burden of NSVT in unclear cases.24 This approach is reasonable. If non-invasive ambulatory monitoring is unrevealing, implantable loop recorders should be considered. Implantable loop recorders may be most useful when a patient has risk factors of unclear clinical significance, such as infrequent symptoms suggestive of an arrhythmia, malignant genetic mutations or imaging findings concerning for fibrosis.
Abnormal Blood Pressure Response to Exercise
As much as 33 % of patients with HCM have an abnormal blood pressure response to exercise.45 An abnormal blood pressure response is a failure to augment or sustain blood pressure while exercising. This is defined as a decrease in systolic pressure from baseline by 20 mmHg during exercise, or a failure to increase systolic pressure by 20 mmHg while exercising. Studies have demonstrated that an abnormal blood pressure response has a very low positive predictive value of 15 %, but a high negative predictive value of 95 % in determining risk of SCD.45,46 If a patient has a dynamic obstruction causing an abnormal response to exercise, the guidelines suggest re-assessing any abnormal blood pressure response after treatment to relieve the obstruction.
Potential Risk Modifiers
Risk modifiers can be used in conjunction with established risk markers to determine a patient’s risk of SCD.24 The risk modifiers are LGE on cMRI, LV apical aneurysm and genetic mutations. LGE is a maker of cardiac fibrosis that is strongly associated with increased ventricular ectopy and NSVT,47,48 and since the guidelines have been written more data have emerged supporting the use of LGE in risk stratification.17,20
Apical aneurysm formation is a rare finding in patients with HCM. The LV apex is dilated and thin-walled with significant scarring, which predisposes these patients to malignant arrhythmias.49 The documented incidence of SCD in these patient is as high as 5 % a year.50 Often these patients present with sustained monomorphic VT, rather than VF, caused by the large apical scar.
While ‘malignant’ genetic mutations of the cardiac sarcomere are discussed in the ACCF/AHA guidelines, the authors suggest that the evidence is poor and routine screening would be of limited value in risk stratification.51
ACCF/AHA Recommendations for Implantable Cardioverter-defibrillator Implantation
The current ACCF/AHA guidelines on ICD implantation are an evolution of the shared ACCF and European Society of Cardiology (ESC) guidelines written in 2003.23 The risk factors listed above were described in the 2003 guidelines, and are used systematically to risk stratify patients in the 2011 ACCF/AHA HCM guidelines. The guidelines also stress the importance of individualised decision-making in determining the need for ICD therapy, as HCM is a heterogeneous disorder with varied phenotypes. Nonetheless, the use of established risk factors as well as risk modifiers can help guide clinicians in selecting patients who would benefit from ICD therapy. The difficulty in using our existing knowledge of SCD in HCM is that our current established risk markers have a low positive predictive value (10–20 %).24,30,52 To improve the predictive value, the guidelines combine certain risk markers with risk modifiers. It should be noted that simply adding risk factors does not always result in a cumulative increase in the risk of SCD.29 A recent study found no increase in ICD intervention rate in patients with multiple risk factors compared to patients with one risk factor.29,52 The current ACCF/AHA recommendations for ICD implantation are summarised below.24
Class I Recommendation
ICD therapy should be recommended to individuals with a documented history of cardiac arrest, VF or haemodynamically-significant VT. There is strong expert consensus that secondary prevention of SCD with ICD implantation is appropriate.24,53 As discussed, this is the highest-risk cohort and will benefit the most from ICD therapy. This recommendation is supported by B level evidence (data derived from non-randomised studies or a single randomised trial).
Class IIa Recommendations
ICD implantation is considered reasonable when patients have any of the following established risk markers: sudden death in one or more first-degree relatives; maximal LV wall thickness ≥30 mm; or a recent and unexplained syncopal event. It is also reasonable in select patients with NSVT, especially those under the age of 30 who have other established risk markers or risk modifiers. ICD implantation can also be considered in patients with an abnormal blood pressure response to exercise in the presence other risk factors or modifiers. The above indications also apply to children. These recommendations are based on C level evidence (consensus opinion of experts, case studies and standard of care).
Class IIb Recommendations
The usefulness of ICD implantation is unknown when patients have only NSVT or an abnormal blood pressure response to exercise in the absence of any other risk marker or modifier.
Class III Recommendations
ICD placement should not be performed in the following scenarios: routine implantation regardless of SCD risk; to allow patients with HCM to participate in competitive sports; and in patients with an identified HCM genotype but no clinical manifestation of HCM.24
European Society of Cardiology’s Hypertrophic Cardiomyopathy Risk Calculator
Historically the North American and European cardiovascular societies have issued a shared guideline regarding ICD implantation for HCM.23 This has recently changed with the 2014 update to the ESC guidelines.53 Unlike the previous shared guidelines and the current ACCF/AHA guidelines, the ESC guidelines advocate the use of a risk calculator to determine an individual patient’s risk of SCD. The scoring model that the ESC risk calculator uses is derived from a large European multicentre cohort of 3,675 patients with HCM.54 In the study, statistical modelling was employed to find clinical variables that were associated with SCD at ≥15 % significance. In the ESC risk calculator, these variables are weighted and produce a score that is expressed as the risk of SCD over 5 years. A score of greater than 6 % over 5 years is considered high risk and indicates that an ICD is recommended; a score of less than 4 % over 5 years is considered low risk and ICD placement is unlikely to be indicated. Unlike the ACCF/AHA guidelines, the ESC risk-scoring algorithm does not treat clinical variables as binary; it assigns relative weight to different variables. This approach may be preferable, as most clinical variables used to determine the risk of SCD have at most modest positive predictive value when evaluated in isolation.
The seven clinical variables that the ESC risk score incorporates are: age, maximum LV-wall thickness, LVOT gradient, left atrial size, NSVT, family history of SCD and unexplained syncope. Some of these variables are not part of the ACCF/AHA guidelines (age, LVOT obstruction and left atrial size). In contrast, some of the risk modifiers that the ACCF/AHA guidelines use, including an abnormal blood pressure response to exercise, are not included in the ESC risk calculator.
The performance of the risk calculator is currently under debate. There is currently evidence favouring and opposing the use of the risk ESC risk calculator when compared to the ACCF/AHA guidelines. The initial external validation studies performed with a cohort of 706 patients found that the ESC model is theoretically better at discriminating low- from high-risk patients.55 A group of 502 patients with HCM in Argentina where studied in a similar fashion. The authors concluded that the ESC risk score accurately categorised all patients who experienced an ICD shock or SCD as either intermediate or high risk.56
There are, however, concerns that the risk calculator has poor sensitivity compared to the ACCF/AHA guidelines. A recent study designed to determine the calibration of the calculator used a cohort of 1,629 patients previously risk-stratified according to the ACCF/AHA guidelines.57 The authors found that the ESC calculator had adequate specificity but poor sensitivity compared to the ACCF/AHA guidelines. Fifty-nine per cent of patients who were stratified as high-risk by the ACCF/AHA guidelines and went on to have an appropriate ICD intervention or SCD would have been classified as low-risk if the ESC calculator had been used. Given the heterogeneity in the molecular and structural abnormalities that characterise HCM, it is especially important that an algorithmic score is validated in diverse populations with varying genotypes and phenotypes. Further validation or revision of the ESC risk calculator will be needed before it can fully supplement the current ACCF/AHA guidelines in North America.
Invasive Testing in Risk Stratification for Implantable Cardioverter-defibrillator Implantation
The use of electrophysiological (EP) testing to risk stratify patients with HCM is controversial. The largest study, performed in the late 1980s, demonstrated a decrease in 5-year survival in patients when VT – either polymorphic or monomorphic – was induced with aggressive programmed ventricular stimulation.58 Studies from this period in time are difficult to apply as most of the patients studied with EP testing would be considered high risk by current guidelines and already have an indication for ICD therapy. The study also required the use of aggressive stimulation protocols requiring right and left ventricular sites in 71 % of all inducible patients. Polymorphic VT was the most commonly induced arrhythmia, occurring in 76 % of inducible patients. Polymorphic VT is a less sensitive marker, as up to one-third of individuals without structural heart disease can develop polymorphic VT with similar stimulation protocols.58,59
Given the unclear indication, as well as potential risk, there is no clear consensus on when to perform EP testing in patients with HCM. There are reported cases of fascicular VT amenable to ablation in patients with HCM. One appropriate use of EP study would be to diagnose and possibly treat patients who present with sustained monomorphic VT and may have a fascicular or bundle branch re-entrant VT mechanism.60,61 Acknowledging the unclear role of EP testing, the AHA/ACC/ESC 2006 guidelines for the management of patients with ventricular arrhythmias issues a class IIb level of evidence (C) for the use of routine EP testing in patients with HCM for the purpose of risk stratification.62
Issues Related to Implantable Cardioverterdefibrillator Implantation
Once a patient is deemed to potentially benefit from ICD therapy, there are several issues to resolve prior to the implantation. They include discussing the risks of ICD-related complications and determining the type of defibrillator system to implant.
ICD-related complications are an important factor to consider in patients with HCM, as many are young and will have a device in place for much of their adult lives. There are two particular risks to discuss in depth with the patient: the risk of device-related complications/ malfunction; and the risk of inappropriate ICD therapy.
Given the variability of age and other comorbid conditions, the risk of ICD complications varies depending on the patient. For example, children and teenagers appear to have a higher incidence of lead fractures due to the strain placed on leads by their growth and development.63 Younger patients will also require multiple ICD generator changes throughout their life, which increases the risk of device-related complications. The published rate of mechanical complications due to ICD implantation is between 4 and 6 % a year in the HCM population.29,64 This includes immediate device complications, such as pneumothorax, pericardial effusion and haematoma or pocket infection. It also includes long-term consequences such as endocarditis and upper extremity venous thrombosis.65
Knowledge of ICD lead performance is growing. Studies suggest that 60–72 % of ICD leads are functioning at 8 years, and that patients who undergo revision have a markedly higher incidence of repeat lead failure or revision.66 This is of particular importance as many HCM patients will require dependable functioning transvenous leads for decades.
Inappropriate ICD therapy, defined as any defibrillation or antitachycardia pacing (ATP) delivered by the device for events other than sustained VT or VF, is another complication related to ICD implantation. A recent analysis described the rate of inappropriate device therapy in a cohort of adult patients with HCM who underwent ICD implantation.29 The authors hypothesised that modern ICD programming, with prolonged detection zones and better supraventricular tachycardia (SVT) discrimination, could lead to a reduction in inappropriate ICD therapy. The authors found that 20 % of patients in the cohort were treated inappropriately despite modern ICD programming. The inappropriate therapy was initiated by atrial fibrillation (39 %), T-wave oversensing (24 %), sinus tachycardia (16 %), atrioventricular nodal re-entrant tachycardia (8 %) and lead fracture (8 %). The rate of appropriate therapy was similar for all patients, regardless of whether they received any inappropriate shock (3.2–3.4 % per year). These findings are similar to other observational studies and mark an avenue for further improvement in current implantation techniques and programming or new device placement strategies.
Implantable Cardioverter-defibrillator Device Selection
Single- or dual-chamber transvenous ICDs are the most common devices that are implanted for primary and secondary prevention of SCD. Single-chamber ICDs are the current guideline-based recommendation for patients with HCM who are at high risk for SCD, but without other compelling reasons for dual-chamber pacing. This is especially true in young patients, who will be exposed to the risks of intra-cardiac leads for an extended period of time. Dual-chamber devices are recommended for patients with other indications for dualchamber pacing, such as pre-existing paroxysmal atrial fibrillation with rapid rates or sinus bradycardia.24 For instance, the older patient with heart failure symptoms and elevated resting outflow gradients (>50 mmHg) may benefit from a dual-chamber system to potentially reduce his or her gradient and heart failure symptoms.24 When compared with single-chamber ICDs, there are no consistent data that dual-chamber devices reduce the likelihood of receiving an inappropriate shock.67
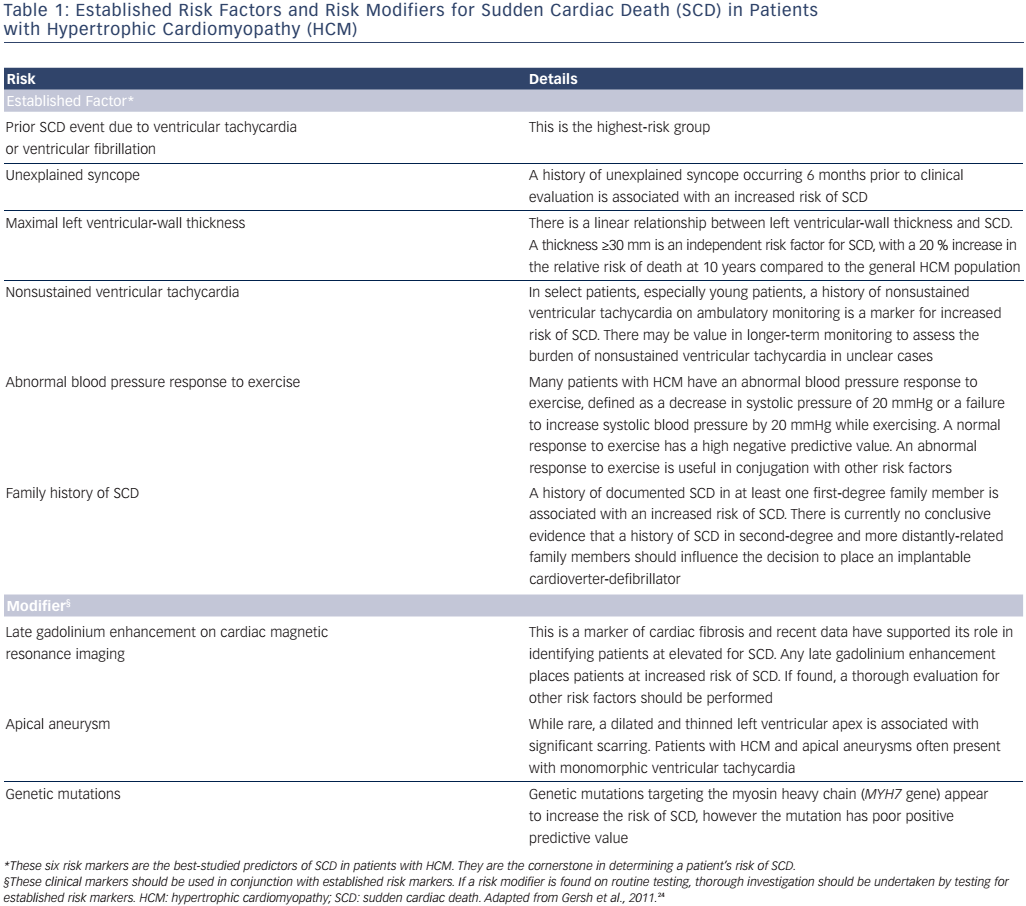
The advent of the fully subcutaneous ICD (S-ICD), see Figure 1, has provided a valuable alternative for certain patients with HCM. It avoids the need for transvenous leads with the entire device implanted below the subcutaneous layer of the chest. The S-ICD allows patients without a need for pacing to have the benefits of arrhythmia protection without the risks of intravascular lead infection or failure. This is particularly useful in young patients with HCM who could avoid the complications associated with transvenous leads as well as the potential procedural risks related to the removal of existing transvenous leads.
To qualify for S-ICD placement, patients undergo a screening procedure to ensure that their QRS and T-wave morphology and amplitude are analysed in various positions. If the T-wave is too large or delayed in relation to the QRS complex, then there is a risk of T-wave oversensing and double counting of the heart rate, which can cause inappropriate therapy.68
The S-ICD appears to be effective in safely terminating VT/VF in patients with HCM. In a single-centre study of 16 patients with HCM, the S-ICD appropriately terminated VF in every patient with 65 J of energy and in 80 % of patients with 50 J. A body mass index >34 was the only baseline characteristic that was associated with an unsuccessful 50 J shock.69 This early success led to long-term followup studies that also appear promising.70
A recent publication of a large pooled S-ICD cohort of 872 patients, including 100 patients with HCM, indicates that the S-ICD can effectively terminate spontaneous life-threatening arrhythmias.71 When examining patients with HCM and S-ICDs, the authors found that 3 % had received appropriate and successful therapies for monomorphic VT. No patient had an episode of VF during the study period. The complication rate of the S-ICD in patients with HCM was low: one patient developed a haematoma, one patient had electrode movement during the procedure, two patients were found to have suboptimal positioning of either their leads or the generator, and two patients developed an infection that required device explantation. The overall infection rate in the HCM subgroup was 1.6 % per year. There were no lead malfunctions in this cohort. The safety data demonstrate low rates of infection and lead disruption in the S-ICD. It is also important to note that, unlike transvenous systems, extraction does not carry the risk of intravascular perforation. Inappropriate therapy occurred in 12.5 % of patients with HCM who had an S-ICD, similar to the non-HCM cohort. T-wave oversensing was the cause of inappropriate therapy in 83 % of HCM patients. This is in contrast to studies of patients with transvenous leads, which have shown SVT to be the main cause of inappropriate therapy.29 SVTs such as atrial fibrillation and flutter are common arrhythmias in patients with HCM.72 In a large study examining the outcomes of transvenous ICD systems, atrial fibrillation/flutter was responsible for 51 % of all inappropriate ICD shocks in patients with transvenous devices.64 In the S-ICD pooled cohort, SVTs (including atrial fibrillation/flutter) were responsible for only 17 % of all inappropriate ICD shocks. Prolonged tachycardia detection time and the S-ICD specific discrimination algorithm may explain why fewer patients with S-ICDs appear to receive inappropriate shocks for SVT.
Avoiding inappropriate device therapy with the S-ICD requires discrimination between VT and a narrow complex tachycardia with a small QRS:T ratio. The advent of dual zone programming is reported to reduce the rates of inappropriate sensing by up to 40 %.73 Patients with HCM may provide a special challenge in avoiding inappropriate shocks due to the progressive nature of hypertrophy and the activity level of younger patients. Both the QRS complex and T-wave may change in amplitude and morphology as the ventricle hypertrophies in HCM.74 This can lead to a mismatch between the template stored at implant and the QRS and T-wave sensed by the device during tachycardia. Young patients with HCM may also exercise vigorously and develop rate-related aberrancy in conduction that can lead to failed SVT discrimination. One option is to have active patients with HCM perform an exercise test prior to implantation in order to apply the ECG template to the ECG during exercise to screen for patients who should not receive the device. It is also helpful for patients who qualify and undergo successful implantation to undergo stress testing after implantation. This will allow the device to store a morphology that can be used when the patient is achieving high heart rates with exercise. It is important to note that while the rate of inappropriate therapy with S-ICD systems needs improvement, it is well within observed rates of transvenous leads.29 Sensing algorithms in the S-ICD have been modified recently to reduce the risk of T-wave oversensing by the device. The problem will likely improve with time as further algorithm refinements are made to avoid T-wave oversensing.
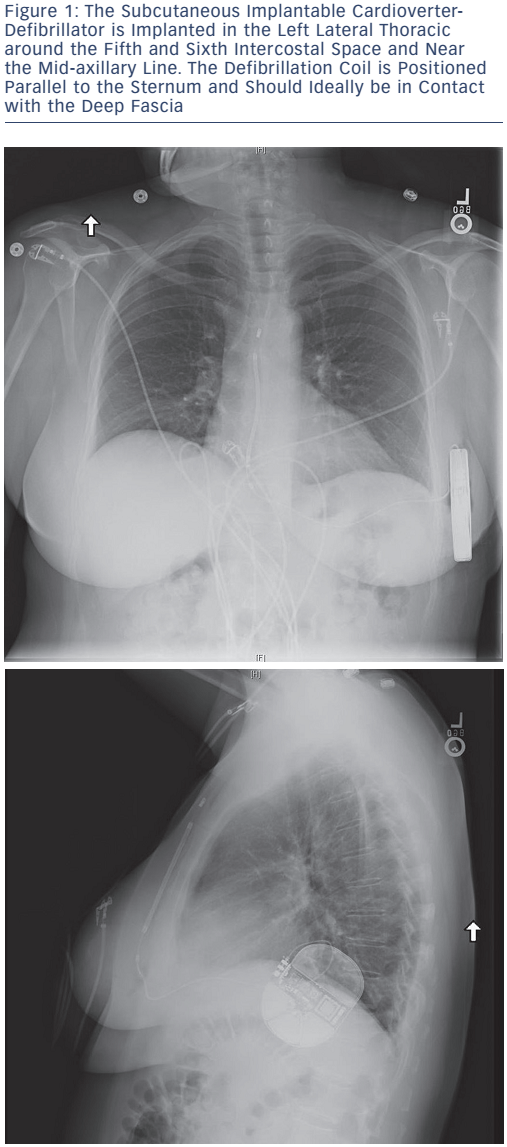
There is concern by some that S-ICDs may not be appropriate for patients with HCM due to the lack of ATP. Monomorphic VT is a common finding in HCM, and ATP therapy has been shown to be modestly successful in terminating monomorphic VT in certain instances.75 Data from Multicentre Automatic Defibrillator Implantation Trial to Reduce Inappropriate Therapy (MADIT RIT) patients who received appropriate ATP therapy showed no reduction in the number of appropriate ICD shocks.76 The modest, if any, benefit of ATP availability does not appear to outweigh the significant risks of complications from transvenous systems. Current data suggest that an S-ICD is as effective at terminating VT/VF in patients with HCM (see Figure 2) as transvenous systems, with a similar rate of inappropriate therapy. A recent, large retrospective trial comparing the clinical outcomes of the S-ICD to transvenous systems in a heterogeneous group of 1,160 patients demonstrated similar trade-offs between the two systems as discussed earlier.77 The authors’ analysis demonstrated that the S-ICD system was more likely to deliver inappropriate therapy due to t-wave oversensing while the transvenous systems were more likely to deliver inappropriate therapy due to SVTs. S-ICD patients had more non-lead related complications which included pocket erosion, defibrillation testing failure and device failure (9.9 % versus. 2.2 % in the transvenous group). The study once again demonstrated a major advantage of the S-ICD: a decrease in lead complications which were defined as replacement or repositioning of the leads (0.8 % vs 11.5 % over 5 years) in the S-ICD group. It is likely that, as S-ICD technology matures and implanters become more comfortable with the device, non-lead related complications will be reduced. The totality of evidence suggests that an S-ICD may be the optimal option in many patients with HCM, especially in young patients who should not be subjected to the risk of transvenous lead failure if it can be avoided.
Clinical Perspective
- Hypertrophic cardiomyopathy is a heterogeneous genetic disorder that increases the risk of sudden cardiac death (SCD). Implantable cardioverter-defibrillator (ICD) therapy is a safe and efficacious means of preventing sudden death in patients with an elevated risk of ventricular arrhythmias.
- There are multiple clinical risk factors to help determine an individual’s risk of SCD. Most clinical variables have excellent negative predictive value but modest positive predictive value. Individualised decision-making is imperative in determining the risk of SCD.
- Traditional transvenous ICD therapy is effective at preventing SCD; however, it is associated with risks such as device infection, lead complications and inappropriate therapy.
- The fully subcutaneous ICD provides the benefits of preventing SCD with a completely extracardiac system. This may be ideal for hypertrophic cardiomyopathy patients, especially younger patients who may have an ICD for most of their adult life.
Conclusion
There has been significant progress in understanding the relationship between sudden death and HCM in the past 50 years. The implantation of an ICD in patients who are deemed to be at high risk of SCD has significantly altered the mortality rate. The current strategy of primary prevention relies on risk stratification based on clinical variables. To improve care for patients with HCM we will need to improve how we identify high-risk patients, perhaps with more refined genetic and imaging data, as well as continuing to refine our treatment options for HCM with devices such as the S-ICD.