







General understanding of early repolarisation (ER) has dramatically changed in the last decade. For several years, ER has been considered a benign electrocardiographic (ECG) finding with high prevalence in the general population. Recently different studies have challenged this view and showed a significant association with life-threatening arrhythmias.1–5
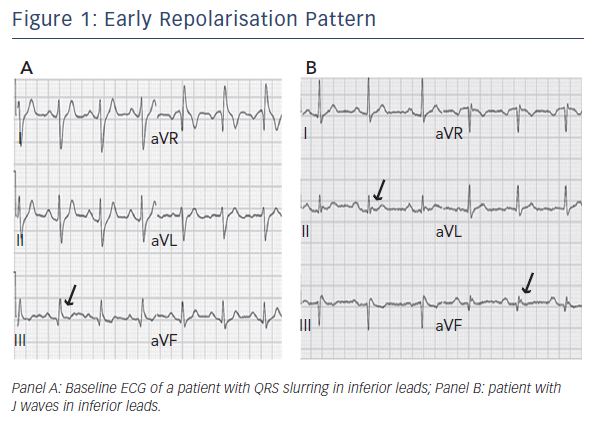
In 2008 Haïssaguerre et al. first reported an increased prevalence of a particular pattern of ER on the resting 12-lead ECG of patients with history of idiopathic ventricular fibrillation (VF) (see Figure 1).2 In these patients, ER was characterised by elevation of the QRS-ST segment junction of at least 0.1 mV above the baseline level, manifesting as QRS slurring (a smooth transition from the QRS complex to the ST segment) or notching (a positive J deflection of at least 1 mm inscribed on the S wave) in two adjacent inferior (II, III and aVF), lateral (I, aVL, and V4–V6), or infero-lateral leads. The ER pattern was observed in 31 % of patients with idiopathic VF and in 5 % of healthy control subjects. Moreover, patients with idiopathic VF and ER pattern presented a higher risk of experiencing an arrhythmic recurrence during a 5-year follow-up.2 This observation was confirmed by a case-control study showing that J-point elevation in the inferior and lateral leads is more frequent in patients with idiopathic VF than in matched control subjects (27 % versus 8 % in inferior leads; 13 % versus 1 % in lateral leads).3 Conversely, ER localised exclusively in V4 to V6 occurs with similar frequency among patients and healthy subjects.3
ER pattern has been shown to be an arrhythmic marker even in the general population of healthy subjects. Tikkanen et al. observed ER pattern in 5.8 % of community-based general population of middleaged subjects.4 Interestingly, healthy individuals with an ER pattern of more than 0.2 mV in inferior leads presented a significantly increased risk of death from cardiac causes and had a higher risk of fatal arrhythmic events.4
A recently published study by Siebermair et al. confirmed that ER pattern in patients with idiopathic VF remains a strong risk factor for arrhythmia recurrence during a very long-term follow-up. Over time, appropriate implantable cardioverter-defibrillator (ICD) interventions can occur in up to 43 % of these patients with idiopathic VF and are observed more often and earlier in patients with ER pattern.5 Based on these findings, when associated with ER pattern, idiopathic VF is considered a distinct clinical entity and has been included in the group of inherited primary arrhythmia syndromes as ER syndrome.6
Notably, ECG definition of ER varies considerably among studies. A recent expert consensus paper has attempted a systematic definition of ER.7 It can be misinterpreted as fragmentation of the QRS complex.7,8 To be considered ‘true ER’, an end-QRS notch or slur should occur on the final 50 % of the downslope of an R-wave. In contrast, fragmentation of QRS complex consists of a notch midway on the downslope of an R-wave. Moreover, ER can present with an ascending ST segment (if amplitude of the ST segment 100 ms after J point termination is greater than amplitude at J termination) or with a descending or horizontal ST segment (if ST-segment amplitude 100 ms after J point termination is less than or equal to the amplitude at J point termination). ER with high amplitude (>0.2 mV) in the inferior leads, associated with a horizontal or descending ST segment, confers the highest arrhythmic risk. Conversely, ascending ST segment has not been found associated with a higher risk of lifethreatening arrhythmias. Finally, ST segment in the absence of a slur or a notch should be considered as nonspecific ST-segment elevation rather than ER pattern.7
Brugada Syndrome and Early Repolarisation
Brugada syndrome (BrS) is an inheritable syndrome characterised by an increased risk of sudden cardiac death (SCD) in patients without overt structural cardiac abnormalities.6,9
The Brugada brothers first described the disease as a new distinct clinical and electrocardiographic entity in 1992.9 The initial report included a series of eight patients presenting with incomplete right bundle branch block, ST-segment elevation on 12-lead ECG and susceptibility to sustained ventricular arrhythmias. All the patients had no electrolytic or ischaemic disturbances nor obvious structural heart disease that could explain the ECG findings.9 Interestingly, an ECG pattern similar to coved-type ST-segment elevation was previously reported as a normal variant in the healthy population or related to VF in patients with structural cardiac abnormality.10,11 Subsequently, a group of Italian researchers considered it as a form of arrhythmogenic right ventricular cardiomyopathy.12 The identification of the first putative casual gene mutation in 1998 clarified the controversy confirming the genetic nature of the disease.13 Over the past two decades a considerable number of studies, including reports of two consensus conferences, contributed to definition of the clinical characteristics, and of cellular and molecular features associated with the disease.14–18
Three different ECG patterns have been identified in patients with BrS (see Table 1). Although all three patterns can be present in BrS and even in the same patient at different times, only type 1 ECG is considered diagnostic of the syndrome.17–20
In fact, according to the last expert consensus document on inherited primary arrhythmia syndromes, BrS is definitively diagnosed when a type 1 ST-segment elevation is observed either spontaneously or after intravenous administration of a sodium channel blocking agent in at least one right precordial lead (V1 and V2), placed in a standard or a superior position (up to the second intercostal space).6 Moreover, in 2012 a group of experts produced a consensus document on ECG criteria outlining a number of new features that can help to identify the Brugada type 1 ECG. According to this document, the ECG pattern, to be considered as type 1, has to display:
- ST-segment elevation with the highest point of QRS-ST of at least 2 mm in lead V1;
- Coved-type morphology (concave or rectilinear, followed by a negative and symmetrical T-wave);
- Progressive decline of the ST-segment morphology (the high take-off is always higher than 40 ms later and this is, in turn, higher than after 80 ms);
- Slow ST-segment descent after the QRS peak (<0.4 mV at 40 ms);
- Ratio of peak height of QRS-ST: peak of ST segment after 80 ms >1
- Greater duration of QRS complex in V1 and V2 than in middle and left precordial leads;
- Location in V1 or V2 but never exclusively in V3;
- Absence of a wide S-wave in lead I and V6.
Moreover, given the minimal morphological differences between type 2 and type 3 and the lack of impact on prognosis, both patterns have been unified in the type 2 ECG.19
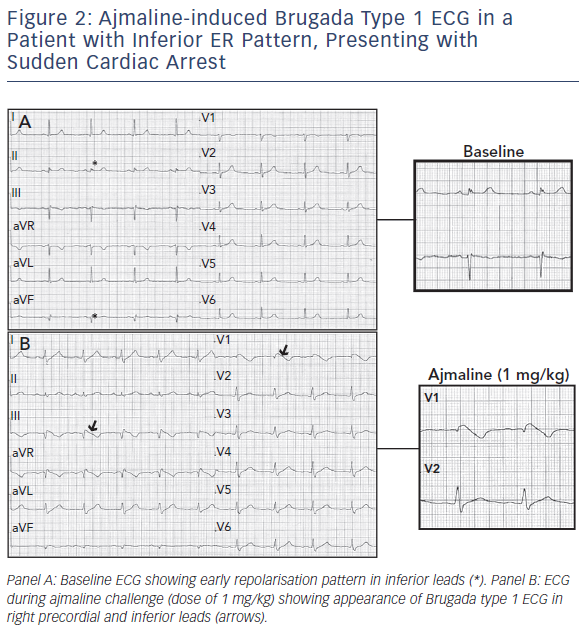
The heterogeneity of BrS can lead some patients to present with additional repolarisation abnormalities such as ER signs in the inferolateral leads (see Figure 2).21,22 Up to 12 % of patients with BrS have the infero-lateral ER pattern. It is more frequently associated with type 1 ECG and previous symptoms, although no significant association with a worse outcome was reported, as shown by a multicentre study.21,22
Patients with BrS experiencing electrical storms have a higher prevalence of ER pattern.23 Moreover, Kawata et al. reported ER pattern in up to 63 % of patients with BrS and documented VF and a worse outcome when ER pattern was persistent.24 It is worth noting that previous studies on ER syndrome have potentially included patients with BrS and nondiagnostic baseline ECG. A very recent study has, in fact, shown that up to 30 % with an initial diagnosis of ER syndrome display spontaneous Brugada type 1 in the high intercostal right precordial leads only, and most VF recurrences occur in patients with ER pattern and Brugada type 1 ECG documented in any of the right precordial leads.25
Based on such findings, sodium channel blocker challenge and ECG recording in high intercostal spaces should be performed in any case of unexplained VF and documented ER pattern in order to exclude the presence of a channelopathy. In BrS, repolarisation signs can coexist with depolarisation abnormalities and a combination of such findings seems to confer an even higher risk of further arrhythmic events.26,27 Tokioka et al. reported a combination of ER pattern and fragmented QRS in 3.6 % of patients with BrS. Both ER pattern and fragmented QRS were identified as independent predictors of further arrhythmic events, and patients with both ERP and fragmented QRS had a significantly higher frequency of arrhythmic events than did those who had neither ER nor fragmented QRS.27
Overlapping Features of Brugada and Early Repolarisation Syndromes
Brugada and ER syndromes are two primary electrical disorders named J-wave syndromes by some investigators.28 Both clinical entities present with distinct ECG abnormalities affecting the junction between the terminal portion of the QRS complex and the beginning of the ST segment. In addition, the two syndromes seem to share a common channelopathy asset that leads to an increased risk of SCD (see Table 1). Antzelevitch and Yan revealed that the presence of an outward shift of balance in repolarising currents in canine wedge preparations, caused by a decrease in sodium or calcium channel currents or an increase in outward potassium currents, creates a notch in the action potential of the epicardium, resulting in a transmural voltage gradient.29 Accentuation of such condition in the right ventricular outflow tract gives rise to BrS coved-type ECG in the right precordial leads; whereas if the infero-lateral ventricle is affected, a J-point elevation, distinct J-wave or end-QRS slur can manifest in the inferior and/or lateral leads.
Brugada and ER pattern can manifest spontaneously or be concealed, becoming apparent only under certain conditions such as fever, high vagal tone, sodium channel blocker challenge (for BrS) or Valsalva manoeuvre (for ER syndrome).30,31 For both syndromes, most prominent ECG changes appear just before the onset of ventricular arrhythmias.2,32 An additional characteristic shared by patients with Brugada and ER pattern is related to the intermittent nature and dynamicity of the ECG pattern over time. Moreover, the ECG phenotype can differ with gender and age categories of patients.33,34 Even the response to ajmaline in BrS can be age dependent.35
The clinical value of repeating ajmaline challenge after puberty in asymptomatic family members with prepubertal negative drug test was recently reported.35 Apart from the ECG, BrS and ER syndrome have several clinical similarities. They present both a highly variable clinical expressivity ranging from a lifelong course to sudden death even in the first months of life.36 Male predominance is a common characteristic.2,33 Moreover, sudden death usually occurs in the third or fourth decade of life.2,36 Ventricular fibrillation is triggered by short-coupled premature ventricular complexes and arrhythmic episodes occur at rest or during sleep. Additionally, they both respond well to quinidine, isoproterenol and cilostazol, explained by the effect of these agents on inhibition of potassium currents or increase of calcium currents.37
Apart from the similar ECG and clinical phenotype, some differences can be also appreciated between the two entities. They include:
- The cardiac region of origin of the ECG phenomena (right ventricular outflow tract in BrS versus inferolateral ventricle in ER syndrome);
- Low-voltage areas (<1.5 mV) with abnormal electrograms present exclusively in the right ventricle of patients with BrS;
- The pro-arrhythmic effect of flecainide in BrS and its effect in attenuating the degree of J-point elevation in patients with ER syndrome;
- The different prognostic role of inducible ventricular arrhythmias at EP study.38–40
In a multicentre study of patients with ER syndrome and aborted sudden death, inducibility of sustained VF during electrophysiology study was relatively infrequent in idiopathic VF survivors (22 %) and did not predict any further arrhythmia during the long-term follow-up.41 In BrS there are many controversies regarding the prognostic role of EP study.42 Although large studies agree that electrophysiological study inducibility is greatest among BrS patients, there has been no consensus on the value of the EP study in predicting outcome.6,43,44 Several consensus documents have addressed this issue and the current recommendation is to implant an ICD in inducible patients (Class IIb indication).6
However, two recent meta-analyses have shown that in BrS, ventricular arrhythmias induced by programmed ventricular stimulation are associated with future arrhythmic events, independently from the symptom status of patients.45,46
One Phenotype: Different Genes
Apart from displaying several clinical similarities, BrS and ER syndrome share a very complex genetic architecture and a far from understood genotype–phenotype interaction.
Brugada and ER syndromes are both genetically heterogeneous and have a considerable allelic heterogeneity: mutations in different genes can lead to the same clinical manifestation and different mutations within each gene can cause the same disease. In addition, different gene mutations and variants can coexist and affect one or more subunits of the potassium, calcium and sodium channel structures.47 BrS has been associated with mutations in 19 different genes, whereas ER syndrome has been associated with mutations in seven genes.
The first gene associated with ER syndrome was KCNJ8, which encodes a pore-forming subunit of the ATP-sensitive potassium channel (Kir6.1-IkATP). The KCNJ8-S422L variant mutation was first described in a young female with ER pattern and frequent episodes of VF.48 Subsequently, loss of function mutations was found in the SCN5A gene and L-type calcium channel genes (LTCC, CACNA1C, CACNB2, CACNA2D1) in patients with idiopathic VF and ER.49,50 Moreover, genetic variants have been identified in the ABCC9 gene, encoding the ATP-binding cassette transporters of ATP-sensitive potassium channels.51 All these gene mutations associated to ER syndrome might enhance the underlying inward–outward current imbalance responsible for accelerated epicardial repolarisation.
Known genes only account for a small proportion of patients.52 Only a small fraction of identified genetic variants has been examined by use of functional expression studies to establish causality or the potential contribution to the pathogenesis of the disease. BrS and ER syndromes can present as familial or isolated cases.53 Malignant familial forms of ER have been reported to be transmitted as an autosomal dominant trait in three large French families.54 Similarly, inheritance in BrS occurs via an autosomal dominant mode of transmission with incomplete penetrance. Most individuals diagnosed with BrS have an affected parent. The proportion of sporadic cases caused by de novo mutation is lower.53 Moreover, the yield of DNA testing in BrS is higher in familial cases (44 %) as compared with isolated cases (21 %).52
After the identification in 1998 of the first gene linked to BrS, the SCN5A gene encoding for the alpha subunit of the cardiac sodium channel, other responsible genes have been reported.13,55 In all genotypes, either a decrease in the inward sodium or calcium current, or an increase of the outward potassium currents has been shown to be associated with the BrS phenotype. Genetic abnormalities are found in up to one-third of genotyped patients, and for the SCN5A gene alone more than 300 mutations have been described.56 Reported mutations include missense mutation, nonsense mutation, nucleotide insertion/deletion and splice site mutation. Loss of function of the sodium channel, which impairs the fast upstroke in phase 0 of the action potential, occurs because of decreased expression of Nav1.5 proteins in the sarcolemma, expression of non-functional channels, or altered gating properties (delayed activation, earlier inactivation, faster inactivation, enhanced slow inactivation and delayed recovery from inactivation).57
The specific type of SCN5A mutation may affect the phenotype. In fact it has been reported that mutations leading to a stop codon, where no sodium channel is created, or missense mutation with >90 % peak sodium current reduction seem to be associated with a poorer prognosis compared with mutations resulting in loss-of-function.58 Reduced sodium current and BrS phenotype can also be due to sodium and calcium channel-associated proteins: GPD1-L, SCN1B and SCN3B. Moreover, loss-of-function mutations in the l-type calcium channel (LTCC) genes encoding for the α and β subunits of the cardiac calcium channel can cause BrS. Putative casual mutations have been also found in genes that regulate transient outward potassium current (KCNJ8, KCNE3, KCND3, KCNE5).
A recent comprehensive mutational analysis of 12 known BrS susceptibility genes in a large cohort of unrelated BrS patients identified SCN5A mutations in 16 %, with the other genes accounting for <5 % of patients.55 The lack of familial segregation data of many susceptibility genes, the relatively frequent association of BrS with common genetic variants and the lack of functional studies remain a major limitation of the genetic testing in BrS.
According to the last consensus document on channelopathies, genetic testing in BrS is not recommended in the absence of a diagnostic ECG. On the other hand, it may be useful and is recommended for family members of a successfully genotyped proband. Sequence analysis of SCN5A should be completed first. If no pathogenic variant is identified, sequence analysis of SCN1B, SCN2B, SCN3B, GPD1L, CACNA1C, CACNB2, CACNA2D1, KCNE3, KCNE1L, KCNJ8, HCN4, RANGRF, SLMAP and TRPM4 may be considered.6
One Gene: Different Phenotypes
Interestingly, all genes related to and potentially involved in the pathogenesis of ER syndrome have been described as associated with BrS.
It has been reported that mutations in the same gene can lead to different phenotypes. As well as BrS, SCN5A mutations may lead to other diseases. SCN5A mutations are implicated in long-QT syndrome type 3, progressive cardiac conduction disease, sick sinus syndrome (or a combination of these), congenital atrial standstill, dilated cardiomyopathy or ER syndrome. A single mutation of SCN5A can lead to several phenotypes in the same family or in a single patient such as BrS, long-QT syndrome type 3, sick sinus syndrome and a variable degree of conduction disturbances (first-degree to complete AV block) known as overlap syndrome.59,60
KCNJ8, LTCC, SCN5A and SCN10A gene mutations have been shown to underline both ER syndrome and BrS. Meideros-Domingo et al. genetically screened 87 probands with BrS and 14 with ER syndrome and found 1 BrS and ER syndrome proband with a S422L-KCNJ8 mutation; the variation was absent in 600 controls.61 Similarly, Barajas-Martinez et al. reported the same missense mutation, p.Ser4222Leu in KCNJ8 in 3 BrS and 1 ER syndrome proband.62
Cases of ER syndrome are often associated with electrical storms in which defects in genes traditionally causing BrS, such as SCN5A, are involved and in combination with mutations in IkATP genes.51 A SCN5A mutation can lead to varying degrees of the Brugada ECG phenotype in members of the same family: in some of them repolarisation abnormalities can occur in the inferior leads only, whereas in others the right precordial leads are characteristically affected.63 This might be related to a different or additional spatial localisation of the affected cardiomyocyte within the heart, other than the right ventricular outflow tract.
A complex genetic inheritance known as the oligogenic model has been recently hypothesised to explain particular clinical presentation of inherited cardiac diseases.47 In contrast to the monogenic paradigm, where a strong monogenic component is responsible of the disease susceptibility, for other diseases such as BrS or ER syndrome inheritance of many genetic risk variants can occur. In addition, the presence of modulating factors may contribute to the manifestation of the disease with a mixed phenotype.64
Conclusion
Brugada and ER syndromes are considered to be two distinct, inherited electrical disorders with overlapping clinical and electrocardiographic features. A considerable number of patients diagnosed with ER syndrome have a genetic mutation related to BrS. Due to its highly variable phenotypic expressivity, patients with BrS may present exclusively with inferolateral repolarisation abnormalities, such as the ER pattern. Moreover, the complex genotype–phenotype interaction in BrS can lead to the occurrence of mixed phenotypes with ER syndrome.
Significant progress in understanding BrS has been achieved since its first description. More than two decades of extensive research on the syndrome have revealed part of its genetic background and electrophysiological and clinical characteristics. The remaining unresolved questions on the genotype–phenotype interaction in BrS provide a stimulus for ongoing active research into the condition. Further functional expression and computational studies will help to elucidate the pathogenic nature and the exact functional consequences of many genetic variants associated with these inherited electrical disorders.