







The incidence of ventricular arrhythmias is often related, within an individual, to the rate of their underlying sinus rhythm (heart rate). The direction of this relationship is generally considered to entail some prognostic significance: whereas ectopic activity suppressed by tachycardia is assumed to be benign, an arrhythmia enhanced by tachycardia is regarded with more concern. Is this assumption valid in general terms? Does it have a mechanistic explanation? To what extent does it apply to arrhythmias occurring in different pathological conditions?
The purpose of this short review is to discuss how arrhythmogenesis, by its diverse mechanisms, can be affected by heart rate.
Effect of Heart Rate on Myocardial Electrical Stability
Heart rate, by itself, has important consequences for at least three factors relevant to electrical stability:
- the pattern of membrane current expression during the action potential;
- intracellular Ca2+ dynamics; and
- cellular energetic competence.
Furthermore, heart rate reflects autonomic balance, which has its own effects on these factors.
Membrane Currents
Membrane current response to tachycardia is mostly relevant to repolarisation and is normally characterised by a net increment of outward (repolarising) current. The main players in this change are an increment in the slow depolarisation-activated K+ current (IKs), whose conductive state accumulates at short diastolic intervals, and a decrease in the high-threshold Ca2+ current (ICaL), which may incompletely recover from inactivation during tachycardia.1 Sympathetic activation, which normally coexists with tachycardia, amplifies rate-induced IKs enhancement, but, to keep up contractile function, it also increases ICaL.2–5 Therefore, both heart rate increment and direct adrenergic modulation of channels contribute to accelerate repolarisation during tachycardia; their concerted action is required for repolarisation stability. Faster repolarisation ensures that propagation velocity and safety (which depend on the availability of the Na+ current) are not compromised by excessive shortening of diastolic intervals. A further consequence of more frequent activation is increased Na+ entry into the cell, which leads to activation of the Na+/K+ pump. The latter carries outward current, which contributes to stabilise diastolic potential, thus preventing its spontaneous depolarisation; this accounts for overdrive suppression of automatic foci.6
Impulse propagation depends on excitation rate only if the diastolic interval is too short to allow for full recovery of the current which supports it (i.e. diastolic interval shorter than refractory period). In normal ventricular myocardium, recovery of the Na+ current (INa) is complete within the end of the action potential; the latter shortens as rate increases, thus making refractoriness rate-adaptive. Therefore, as indicated by constancy of QRS duration over the whole range of physiological rates, propagation is largely independent of heart rate.1 INa recovery may be delayed beyond full repolarisation (post-repolarisation refractoriness) by Na+-channel blockers (Class IC in particular)7 and by loss of diastolic polarisation; furthermore, when a small gap of unexcitable tissue is included in the propagation path, refractoriness may become independent of action potential duration and even acquire an opposite rate-dependency.8 Rate-dependency of conduction (e.g. in bundle branch block) may appear in such conditions; however, it is not necessarily present under conditions of conduction impairment in ventricular myocardium. For instance, in severely fibrotic porcine hearts subjected to hypokalemia, ventricular conduction was depressed, but rate-dependency occurred only above 240 BPM.9 Because of its less polarised diastolic potential and specific INa-gating properties,10 atrial myocardium may be more prone than ventricular myocardium to develop post-repolarisation refractoriness and, thus, conduction rate-dependency;1 however, it is unclear whether this may occur within a physiological range of rates. Post-repolarisation refractoriness and conduction rate-dependency are instead typical of nodal tissue, in which propagation is supported by a Ca2+ current (ICaL) with slow recovery kinetics. However, sinus tachycardia is physiologically induced by sympathetic activation, which enhances ICaL; therefore, unless ICaL conductance is reduced (e.g. by drugs), even nodal conduction is largely rate-independent. To summarise, considering rate-dependent conduction slowing as a general factor in tachycardia-induced arrhythmias may, in our view, be incorrect.
Bradycardia prolongs repolarisation, on the other hand. Longer repolarisation is intrinsically more labile to perturbations11,12 and, as such, prone to become spatially inhomogeneous and temporally variable.13 Moreover, repolarisation velocity is pivotal in preventing partial ICaL recovery and its immediate reactivation during the action potential plateau, which would result in major perturbation of the action potential profile (early-afterdepolarisations, EADs).14 Prolonged repolarisation, either primary or acquired, is notoriously associated with proarrhythmia. While EADs represent the most obvious arrhythmia-initiating event, even in the absence of EADs the tendency of prolonged repolarisation is to be more variable, both in space (repolarisation dispersion or heterogeneity) and time (repolarisation variability). This may lead to the steep voltage gradients, likely setting the stage for local conduction block, which characterise the sites of onset of life-threatening rhythms.15 Fortunately, bradycardia is normally associated with low adrenergic activity, which helps to maintain inward and outward current components in balance.
Overall, changes of membrane current expression during the action potential are such as to confer electrical stability in the case of tachycardia, but much less during bradycardia. Nonetheless, heart rate changes are pivotal in cardiovascular adaptation to metabolic needs; therefore, we can assume that normal myocardium is rigged to face wide heart rate changes. This is conceivably achieved through concerted modulation of sinus node pacemaker rate and of the function of working myocytes.
Intracellular Ca2+ Dynamics
Heart rate has profound effects on intracellular Ca2+ dynamics, which contribute to adapt contractility to the changes in cycle duration. Tachycardia increases the Ca2+ influx/efflux ratio, thereby increasing cell Ca2+ content.16 Adrenergic stimulation contributes to this by enhancing Ca2+ influx (through ICaL) and by stimulating a Ca2+ pump (SERCA2a), which supports fast uptake of the ion by the sarcoplasmic reticulum (SR).16 Under normal conditions, cell Ca2+ content, especially in the SR, is tightly controlled by a complex of feedback mechanisms, which keep it near a set point.17 While the joint effects of tachycardia and adrenergic activation move the set point to increase the Ca2+ available to trigger contraction, the feedback system guarantees achievement of a new steady-state and prevents progressive Ca2+ overload.18 This set of mechanisms ensures SR stability, i.e. capability of the intracellular store to release Ca2+ only in response to the appropriate trigger (the action potential).19 Nonetheless, tachycardia and adrenergic stimulation are undoubtedly stress conditions for the Ca2+ handling system: if any of its components is abnormal, as occurs in common primary and acquired diseases, SR instability is more likely to ensue at high heart rate.20,21 Thus, the appearance of arrhythmias during sinus tachycardia per se, or as a reporter of sympathetic activation, may suggest mechanisms related to abnormality of the Ca2 -handling machinery.
Intracellular Ca2+ dynamics are relevant to arrhythmogenesis because cytosolic Ca2+ ‘talks’ to membrane potential through Ca2+ sensitive mechanisms.19 For instance, due to its stoichiometry, the Na+/Ca2+ exchanger moves a positive charge in the inward direction whenever extruding a Ca2+ ion from the cell; a rise in cytosolic Ca2+ fuels the exchange, thus resulting in membrane depolarisation. Also, the speed of ICaL inactivation, which affects the amount of inward current during repolarisation, strongly depends on formation of a Ca2+/calmodulin complex at the inner mouth of the channel.22 Such a crosstalk translates variability of intracellular Ca2+ dynamics into variability of repolarisation,23,24 with consequences of relevance for arrhythmogenesis. SR instability has a pivotal role in multiple arrhythmogenic mechanisms, including DADs, EADs, repolarisation alternans, and repolarisation variability in general.19,21,23,24
Energetic Competence
Heart rate is the major determinant of oxygen consumption by both chemical (mainly Na+ and Ca2+ pumping) and mechanical (sarcomere shortening) components of cardiac work. To cope with it, mitochondrial production of reduced substrates fuelling the electron transport chain (i.e. nicotinamide adenine dinucleotide) is tightly controlled by mitochondrial Ca2+, whose concentration increases during tachycardia as it grossly follows that of cytosol.25,26 Under these conditions, inadequate availability of O2 (the electron acceptor) paradoxically increases mitochondrial production of reactive oxygen species (ROS), likely because of an imbalance between their generation and scavenging, and reduces ATP generation. ROS-induced proarrhythmia is well known: SR instability induced by peroxidation of "ryanodine receptor" channels has a strong proarrhythmic potential, but dysfunction of other ion channels (e.g. enhancement of the late Na+ current) is also involved.27–33 Furthermore, a drop in the ATP/ADP ratio activates the sarcolemmal ATP-sensitive current (IKATP), which may dramatically shorten repolarisation and impair cell excitability locally,34,35 resulting in marked electrical heterogeneity. Therefore, in the context of ischaemic heart disease, the immediate proarrhythmic potential of tachycardia is obvious; indeed, tachycardia-induced arrhythmias may represent an ischaemia equivalent.
Is the Relation Between Arrhythmia and Heart Rate Relevant to Prognosis and Therapeutic Strategy?
Arrhythmia Facilitation by Tachycardia
In light of the arguments above, whereas tachycardia may afford greater electrical stability in the normal heart, it may well be an arrhythmia trigger when an abnormal substrate is present. The latter may be provided by extremely common conditions, such as ischaemic heart disease, or rarer but very serious ones, such as ryanodine receptor dysfunction of genetic origin (as in catecholamine-induced polymorphic ventricular tachycardias).36 This, together with the generally benign nature of overdrive-suppressible enhanced automaticity (parasystolic rhythms), might lead to the conclusion that ectopic beats manifesting selectively at low heart rates should not be associated with the substrate required for their degeneration into complex, life-threatening arrhythmias.
Arrhythmia Facilitation by Bradycardia
A substantial body of evidence indicates that, contrary to the view above, pronounced bradycardia may also facilitate life-threatening arrhythmias, as predicted by its destabilising effect on repolarisation. Susceptibility to torsade de pointes (TdP) ventricular tachycardias, the type of arrhythmia more specifically linked to repolarisation instability, characterises experimental models of bradycardia37 and may be observed in patients with AV block resulting in low ventricular rates.38 The question is then whether low heart rate is by itself responsible for arrhythmogenesis. In dogs with chronic AV block (CAVB), an extensively characterised proarrhythmia model, bradycardia may trigger TdP only after inducing myocardial remodelling:39 a set of functional and structural myocardial modifications resulting from chronic adaptation to low heart rate. Repolarisation of remodelled myocardium is generally characterised by downregulation of outward currents and upregulation of inward ones, resulting in slightly prolonged action potential duration and reduced repolarisation reserve.40 This experimental observation is matched by clinical data showing that, among AV block patients, those developing TdP are characterised by longer QT intervals and specific features in repolarisation profile.38 Therefore, as in the case of tachycardia-induced arrhythmias, arrhythmia ensuing during bradycardia may indicate the presence of a substrate, represented in this case by reduced repolarisation reserve. Notably, such a substrate may result from myocardial remodelling of whatever aetiology, thus making pronounced bradycardia a concern in all conditions of myocardial hypertrophy/failure.
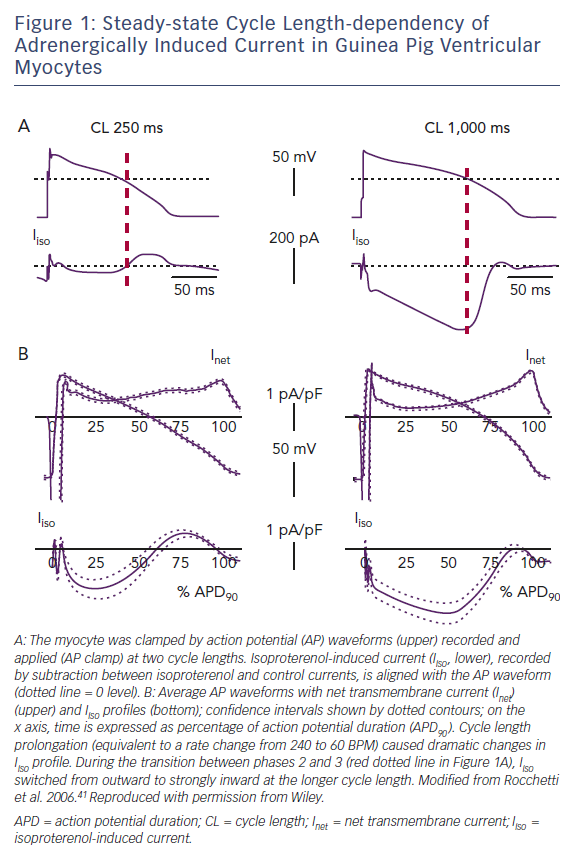
A special condition, sensitising repolarisation to the destabilising effect of bradycardia, may be provided by a mismatch between sinoatrial and ventricular responses to sympathetic activation. Action-potential clamp (AP-clamp) experiments in guinea pig ventricular myocytes showed that the composite current activated by beta-adrenergic receptors (largely IKs + ICaL) switched from partly outward to entirely inward when the pacing rate was reduced.41 Notably, at slow rates the excess inward (depolarising) current impinged on the transition between action potential phases 2 and 3, i.e. when EADs commonly occur (Figure 1).
Bradycardia-induced distortion of the current profile in these experiments was so remarkable to suggest that profound repolarisation abnormalities should be a necessary consequence of inadequate ventricular rate response to sympathetic activation, even when the currents are intrinsically normal. Evidence that this is fortunately not the case in vivo (in the CAVB dog and AV block patients mentioned above) may be partly explained by specificities in the guinea pig action potential,42 but also by failure of membrane current to feedback on membrane potential course under AP-clamp conditions. In other words, under physiological conditions, current distortions would produce changes in the AP profile that may minimise their effect. Nonetheless, the AP-clamp experiments do show that subnormal rate response to sympathetic activation may impose a very significant stress on repolarisation reserve.
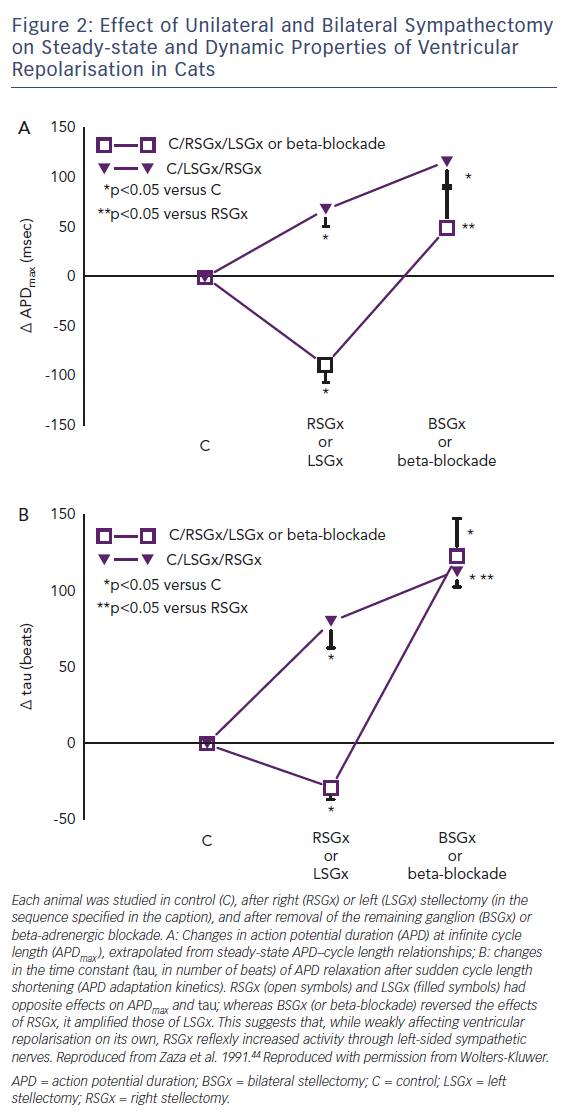
Before the role of genetic channel abnormalities was recognised, long QT syndrome was experimentally reproduced in normal hearts by an imbalance between the activities of left- and right-sided sympathetic nerves.43 Whereas the effects of left stellectomy (i.e. sympathyectomy) on static and dynamic aspects of ventricular repolarisation were similar to those of bilateral denervation, right stellectomy had opposite effects (Figure 2).44 Notably, whereas sinus rate is mostly under the influence of right-sided nerves, left-sided ones prevail in the control of ventricular electrophysiology.43 The concept of "inadequate rate response" introduces a quantitative argument in the discussion of the correlation between heart rate and arrhythmias. For instance, in long QT syndromes, cardiac arrest may be preceded by an increase in heart rate,45 as expected when arrhythmias are triggered by sympathetic activation. However, because of prolonged repolarisation and, in the case of IK deficits, also because of defective sinus automaticity, long QT syndrome patients are intrinsically bradycardic, with subnormal rate response to sympathetic activation,46 the latter being a likely contributor to the genesis of TdP. This has, of course, therapeutic implications in the potential benefits of positive chronotropy, by pacemakers or drugs, under conditions of repolarisation instability.
In conclusion, from the mechanistic viewpoint, when considering the relation between heart rate and arrhythmias, the question should be ‘how appropriate is sinus rate to autonomic balance?’, rather than ‘is heart rate high (or low)?’.
The Importance of Dynamics
We have thus far considered tachycardia and bradycardia as static conditions; nonetheless, emerging evidence indicates that the link between heart rate and arrhythmogenesis may also reside in the dynamics of transitions between different heart rates. This argument is well illustrated by examples, again in the field of repolarisation syndromes.
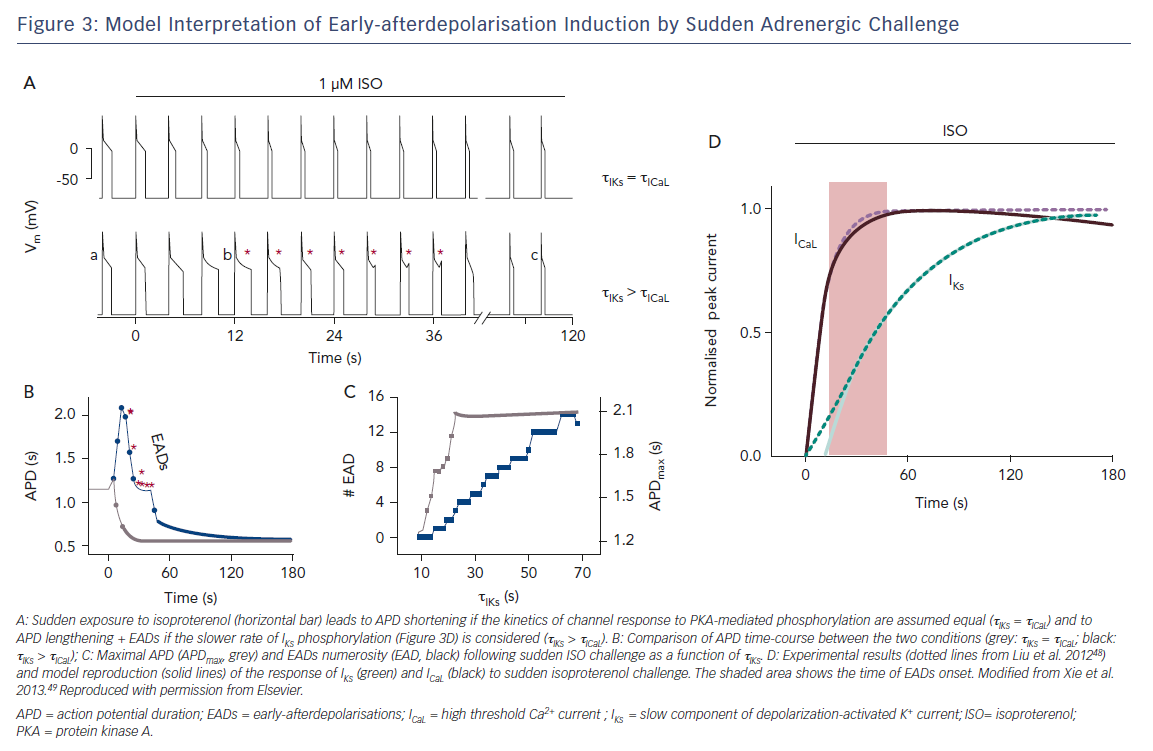
Cardiac arrest in patients with prolonged repolarisation may occur upon sudden emotional stress (e.g. an alarm going off), rather than during sustained physical exercise; this is especially the case for IKr defects.47 This has been elegantly interpreted by showing that ICaL response to an adrenergic surge is considerably faster than IKs response, thus facilitating EADs (Figure 3).48,49 Thus, even if a suitable balance may be preserved during sustained exercise, a dangerous mismatch between depolarising and repolarising currents may occur during sudden sympathetic activation. While the case of prolonged repolarisation syndromes is the most obvious example, sudden transitions between states of vagal and sympathetic activity, and the resulting heart rate changes, may contribute to the high incidence of arrhythmic events during deep sleep stages, particularly when associated with ventilation abnormalities (a powerful trigger of autonomic reflexes).50
In patients with genotype LQT1 of long QT syndrome, membrane current response to adrenergic activation is unbalanced due to primary IKs deficit, thus making these patients particularly susceptible to sympathetic activation, especially with sustained exercise.47 However, the search for concomitant factors affecting the arrhythmic risk has led to the unexpected observation of a higher baroreceptor sensitivity (BRS) in symptomatic versus asymptomatic mutation carriers (still associated with slightly higher heart rate).51
Since high BRS is held to reflect parasympathetic dominance (and found to be protective under other conditions), the finding is surprising, and leads to consider other mechanisms in the contribution of rate changes to cardiac arrest. It has been suggested that high BRS implies brisker autonomic and heart rate changes during haemodynamic perturbations.51 IKs is among the main factors underlying APD adaptation to sudden rate fluctuations: the stiffer repolarisation typical of LQT1 might decrease the tolerance to these fluctuations.52 Indeed, stiffer repolarisation is the sole mechanism we found to potentially account for the severe electrical instability in a calmodulin mutation (F142L) associated with QT prolongation.53 Further investigation is required to confirm this view; nonetheless, these findings highlight the possibility that arrhythmogenesis may also result from a mismatch between the dynamicity of repolarisation and that of heart rate.
Conclusion
With the exception of parasystolic rhythm, in which appearance at slow rates is likely due to relief from overdrive suppression of an automatic focus, attribution of prognostic significance to arrhythmia relationship with heart rate should carefully consider the substrate likely present in the individual patient.
The same is also true for deciding whether heart rate increase or decrease may be a logical antiarrhythmic approach, or can be safely tolerated as a therapy side-effect. In many cases this may be straightforward (e.g. bradycardia-induced arrhythmias in a long QT syndrome patient, or tachycardia-induced arrhythmias clearly related to ischaemia); however, other cases may be more ambiguous. For instance, in heart failure, arrhythmia facilitation by tachycardia may suggest SR instability (or energetic incompetence); however, lowering heart rate in a likely remodelled context may conceivably carry some risk, particularly if adrenergic activation remains unopposed.
Clinical Perspective
- Heart rate may affect electrical stability per se, or reflect autonomic balance.
- Depending on the substrate, arrhythmias can be facilitated by either tachycardia or bradycardia.
- Preferential occurrence of an arrhythmia during bradycardia may imply low risk only if the features typical of parasystole can be detected.
- Inadequate heart rate response to sympathetic activation may contribute to electrical instability.
- Sudden changes in heart rate and/or autonomic balance may contribute to electrical instability.